

- 1.0 Introduction: Why biotech events in Lisbon in 2026 Will Determine the Commercial Success or Failure of the Next Generation of Cutting-Edge Biopharmaceuticals?
- 2.0 Topic 1 at biotech events: The Clash Between Small Nucleic Acids and In Vivo CAR-T—The Decisive Battle Between Extrahepatic Targeting and Novel LNPs
- 3.0 Topic 2 at biotech events: The “Endgame of Hypercompetition” Between Peptides and GLP-1—The Battle Between “Once-Monthly” Microsphere Technology and Highly Absorbable Oral Formulations
- 4.0 Topic 3 at biotech events: The Safety Ceiling of ADCs—Timed and Site-Specific Cleavage of Linkers and In Vivo Circulatory Stability
- 5.0 Cross-Industry Brainstorming at biotech events (Cross-pollination): When ADCs Meet Nucleic Acids and Peptides—The Convergence of DDS Technologies
- 6.0 On the Ground in Lisbon: A Practical Guide to Attending biotech events (and Online Tracking) CRS 2026
- 7.0 Conclusion from biotech events: DDS Is No Longer a Supporting Role; It Is the Crown Jewel of the Biopharmaceutical Value Chain Post-2026
- 8.0 biotech events FAQ: Five Frequently Asked Questions About In Vivo CAR-T, DDS, and the CRS 2026 Annual Meeting
1.0 Introduction: Why biotech events in Lisbon in 2026 Will Determine the Commercial Success or Failure of the Next Generation of Cutting-Edge Biopharmaceuticals?
1.1 Paradigm Shift: The Biggest Upheaval in the Biopharmaceutical Industry in Q2 2026
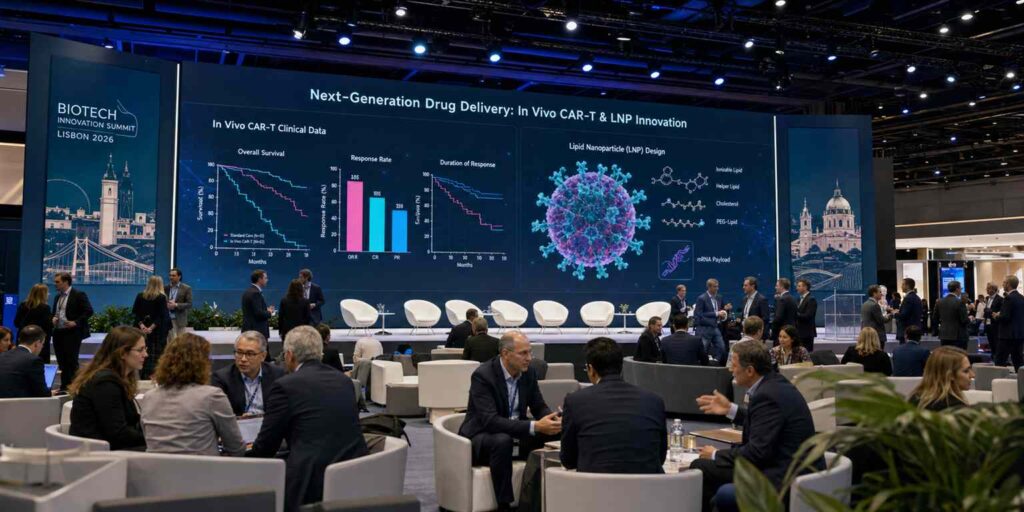
1.1.1 Milestone Developments by Legend Biotech and Boehringer Ingelheim (BI) in the Field of In Vivo CAR-T
On June 3, 2026, at one of the year’s most impactful biotech events, Legend Biotech presented a set of data at the European Hematology Association (EHA) Annual Congress—the first-in-human clinical results of in vivo CAR-T therapy in patients with relapsed/refractory diffuse large B-cell lymphoma (r/r DLBCL).To grasp the significance of these data, you must first set aside everything you’ve known about CAR-T over the past decade. This is no longer the complex process that required extracting a patient’s T cells, shipping them hundreds of kilometers to a GMP cleanroom, painstakingly engineering them with lentiviral vectors for two weeks, and then transporting them back to the hospital via cold chain.This time, Legend Biotech’s approach is shockingly simple: they encapsulated mRNA encoding a CD19 CAR within lipid nanoparticles (LNPs) and administered it directly into patients’ veins via intravenous injection, much like a vaccine. Once in the bloodstream, the LNPs encounter the patient’s T cells, are internalized, and release the mRNA, causing the T cells to transform into CAR-T cells right on the spot—no in vitro manipulation, no lengthy wait, and no manufacturing costs in the millions.
Preliminary data show that among the six evaluable patients in the lowest-dose group, four achieved objective response (ORR 67%), with two of those achieving complete remission (CR 33%).If you have even a basic understanding of cell therapy, you’ll know that for patients with refractory lymphoma who have relapsed or progressed after multiple lines of treatment, this absolute remission rate isn’t particularly impressive—the best ORR for ex vivo CAR-T in this indication can exceed 80%.But the crux of the matter has never been how impressive these data are in and of themselves, but rather that they demonstrate a fundamental principle significant enough to rewrite the textbooks: delivering mRNA via LNP to generate functional CAR-T cells in situ within the human body—this technological pathway is viable.
On the very same day Legend Biotech released its data, German pharmaceutical giant Boehringer Ingelheim (BI) dropped an even bigger bombshell: it announced the acquisition of a biotechnology company specializing in an mRNA-LNP in vivo CAR-T platform for an upfront payment of $450 million and a total transaction value exceeding $2 billion.This is no ordinary deal, as the indication BI is targeting has taken everyone by surprise—not lymphoma, not leukemia, but systemic lupus erythematosus (SLE). SLE is an autoimmune disease with a massive patient population, affecting approximately 5 million people worldwide and over 1 million in China.Existing standard treatments—corticosteroids, immunosuppressants, and belimumab—are either associated with significant side effects or offer limited efficacy. BI’s rationale is straightforward: if in vivo CAR-T therapy can use CD19 CAR to eliminate all B cells producing autoantibodies, it would effectively give SLE patients a chance to “reset” their immune systems.A 2021 academic paper from the University of Erlangen in Germany, which used ex vivo CAR-T therapy to treat SLE, has already validated this concept—all five patients with refractory SLE achieved drug-free remission following infusion of CD19 CAR-T cells. Now, BI aims to use LNP delivery technology to reduce the ex vivo CAR-T’s astronomical price tag—currently in the millions of dollars—to a level affordable for the average person. This is the true business rationale behind the $2 billion deal.
These two milestone events occurred just 24 hours apart, yet they point to the same underlying logic: by 2026, the biopharmaceutical industry has shifted entirely from the frenzy of discovering the next PD-1 to an engineering battle focused on “how to deliver drugs precisely, safely, and cost-effectively into target cells.” And the central battlefield of this campaign is none other than the Drug Delivery System (DDS).
Shortly thereafter, on June 5, Alnylam Pharmaceuticals, a leader in the small interfering RNA (siRNA) field, released Phase II clinical data for its ANGPTL3-targeting siRNA drug in patients with hypertension and hyperlipidemia.The data was remarkable: following a single subcutaneous injection, the reduction in low-density lipoprotein cholesterol (LDL-C) remained above 55% for over 18 months. What does this mean? It means that patients need only two injections per year to achieve the lipid-lowering effect that statins can barely achieve with daily dosing—and this was accomplished without any new safety signals observed regarding liver or kidney toxicity.
Meanwhile, Arrowhead Pharmaceuticals disclosed preclinical data demonstrating central nervous system (CNS) delivery using a novel ligand-conjugation technology—a new chemical linker that covalently binds antibody fragments to siRNA.In mouse and non-human primate (NHP) models, researchers observed for the first time that siRNA molecules, after crossing the blood-brain barrier (BBB), achieved over 70% target gene silencing efficiency in neurons, with effects lasting more than six months.This marks a watershed moment in the history of small nucleic acid drug development—for the past fifteen years, siRNA has been confined to the liver, a single organ; now it has finally “grown legs” and is making its way to tissues such as the CNS, muscles, and lungs, which were previously considered off-limits for small nucleic acids.
| Timeline | Event | Key Implications | Impact on DDS |
| June 3, 2026 | Legend Biotech Announces Phase I Data on In Vivo CAR-T Lymphoma | In vivo in situ generation of CAR-T cells is feasible in humans | LNP-mediated T-cell delivery pathway clinically validated |
| June 3, 2026 | BI Acquires In Vivo CAR-T Platform for $2 Billion | Autoimmune Diseases Emerge as the Final Battleground for In Vivo CAR-T | Top pharmaceutical companies reassess the commercial value of the LNP platform |
| June 5, 2026 | Alnylam Announces Phase II Data for ANGPTL3 siRNA | 18-Month Long-Acting Lipid-Lowering Effect Establishes Overwhelming Advantage in the Small Interfering RNA (siRNA) Chronic Disease Field | GalNAc-conjugation technology has reached commercialization standards |
| June 5, 2026 | Arrowhead Discloses Preclinical Data on CNS Delivery | siRNA Crosses Blood-Brain Barrier in NHPs for the First Time | Novel ligand-conjugation technology opens up the field of extrahepatic targeting |
1.1.2 The Ultimate Leap from “Factory-Scale Cell Therapy” to “In Vivo On-Site Modification”
To understand why in vivo CAR-T represents not just a routine technological iteration but an industry-shaking revolution, you first need to grasp one key figure: just how expensive, time-consuming, and complex ex vivo CAR-T truly is.
Every step of the traditional ex vivo CAR-T production process involves a mounting accumulation of time, money, and risk: drawing blood from the patient’s arm → isolating T cells → cold-chain transport to a GMP cleanroom(often hundreds of kilometers away) → lentiviral vector transduction (inserting the CAR gene into the T-cell genome) → expansion of T-cells to billions in bioreactors → 7 to 14 days of quality control and release → return transport via cold chain to the hospital → the patient undergoes 3 days of lymphodepletion chemotherapy → finally, infusion.This entire process takes anywhere from 3 to 5 weeks. For patients with certain aggressive hematologic malignancies, this waiting period can be fatal—real-world data shows that approximately 15%–20% of patients with advanced lymphoma lose their chance of treatment due to disease progression or complications while waiting for CAR-T preparation.
Then there is the cost. Currently approved CAR-T products, such as Kymriah and Yescarta, are priced between $370,000 and $470,000 in the United States. When combined with costs for hospitalization, pre-treatment, and complication management, the total treatment cost easily exceeds $1 million. CAR-T pricing in China is relatively lower—Legend Biotech’s Carvykti is priced at approximately 1.2 million RMB in China—but it still far exceeds the financial capacity of the vast majority of Chinese families.Progress in medical insurance negotiations has been slow, and the coverage of municipal public health insurance plans remains limited—as of Q2 2026, fewer than 20 cities nationwide had included CAR-T reimbursement in their public health insurance programs. This means that, over the past decade, ex vivo CAR-T has essentially remained a treatment model “reserved for a select few.”
In vivo CAR-T overturns this fundamental logic. First, the production cycle is effectively eliminated.No in vitro procedures, no cell expansion, no quality control and release—theoretically, a patient can go from diagnosis to treatment within the same week. Second, production costs have plummeted. A single bioreactor, the operating costs of a GMP cleanroom, and a team of trained cell preparation technicians—these major cost drivers of ex vivo CAR-T are virtually eliminated in in vivo CAR-T.According to industry analysis reports, the cost per treatment for in vivo CAR-T is expected to be kept under $50,000—just one-tenth to one-twentieth of the cost of ex vivo CAR-T. This is no longer a privilege reserved for the few, but a treatment model with population-level accessibility.
More importantly, in vivo CAR-T does not require patients to undergo lymphodepletion chemotherapy. The purpose of lymphodepletion is to “make room” for the reinfused CAR-T cells—by killing a large number of the patient’s endogenous lymphocytes, thereby reducing competition and rejection of the newly introduced CAR-T cells. However, this process itself is a significant source of toxicity, leading to severe immune deficiency in patients and increasing the risk of infection.In vivo CAR-T therapy circumvents this issue: since CAR-T cells are generated in situ within the patient’s own lymphatic system, there is no need for “clearing the field.” This directly reduces the incidence of treatment-related serious adverse events (SAEs)—in Legend Biotech’s Phase I data, there were no cases of Grade ≥3 cytokine release syndrome (CRS) or immune effector cell-associated neurotoxicity syndrome (ICANS).
| Comparison Dimensions | Ex Vivo CAR-T | In Vivo CAR-T | Commercial Disruptiveness |
| Production Cycle | 3–5 weeks (including shipping, expansion, and quality control) | 0 weeks (ready-to-use injection) | From custom-order to off-the-shelf availability |
| Treatment Cost | $370,000–$470,000 (US) / 1.2 million RMB (China) | Estimated <$50,000 (per treatment) | Accessibility Expanding from the ultra-wealthy to the general public |
| Production Facilities | GMP cleanrooms + bioreactors + specialized technicians | Standardized LNP formulation + standard cold chain | Manufacturing barriers reduced from cell-based to small-molecule drug levels |
| Cytostatic chemotherapy | Mandatory (increases toxicity and infection risk) | Not required | Significantly improved safety, lower treatment barriers |
| Expanded indications | Limited to hematologic malignancies (ineffective for solid tumors) | Oncology + autoimmune diseases + age-related conditions (platform-based) | Total Addressable Market (TAM) expands by more than 10 times |
| High reliance on cold chain | Extremely high (ultra-low temperature + live cell transport) | Moderate (relatively higher LNP stability) | Logistics costs reduced by an order of magnitude |
1.2 Hidden Bottlenecks: A Common Challenge for Cutting-Edge Formulations—“If It Can’t Be Delivered, How Can We Talk About Efficacy?”
1.2.1 The Core of In Vivo CAR-T Is Not Gene Editing, but the Breakthrough in Non-Viral Vectors (LNPs)
If you think the core technological breakthrough for in vivo CAR-T lies in CAR structural design or the precision of CRISPR gene editing, you’re only seeing half the story. The key technology that truly determines whether in vivo CAR-T can transition from academic papers to commercialization is that seemingly unremarkable “delivery vehicle”—the lipid nanoparticle (LNP).
Why is LNP so difficult to develop? Because it must simultaneously fulfill three conflicting tasks within the human body. First, it must remain stable in the bloodstream and not be cleared by the liver, spleen, or kidneys before it finds T cells. Human plasma contains large amounts of nucleases (RNases), and if mRNA molecules are exposed in the blood, their half-life is only a few seconds to a few minutes.LNPs must act as a “human shield,” tightly encapsulating the fragile mRNA to resist degradation by RNases. Second, they must precisely locate the target cells—in this case, T cells—and be effectively taken up by them. But here’s the problem: traditional LNPs (such as those used in COVID-19 mRNA vaccines) inherently exhibit “liver tropism.”Once LNPs enter the bloodstream, plasma proteins rapidly bind to their surface, forming a “protein corona,” with apolipoprotein E (ApoE) as the primary component. Since liver cells have a high density of ApoE receptors (LDLR) on their surface, this causes over 80%–90% of LNPs to be “intercepted” by the liver, preventing them from reaching the lymphoid organs where T cells reside.
Third, even after being taken up by T cells, the LNP cannot remain trapped within the “endosome”—a small intracellular compartment. After entering the cell via endocytosis, the LNP is enclosed within a membrane-bound vesicle called an “endosome.” If the mRNA cannot escape this vesicle, the entire effort is in vain.The ionizable cationic lipids in the LNP must undergo protonation in the acidic environment of the endosome (pH approximately 5.0–6.5). This allows them to interact electrostatically with the anionic lipids on the endosomal membrane, forming a non-bilayer lipid phase. This process disrupts the integrity of the endosomal membrane, allowing the mRNA to escape into the cytoplasm.This process is called “endosomal escape.” Don’t underestimate this step—even the best LNP formulations currently available have an endosomal escape efficiency of only 1%–4%. In other words, out of 100 mRNA molecules taken up by T cells, only 1 to 4 ultimately reach the cytoplasm and are translated into CAR proteins. The remaining 96–99 are all sent to the lysosomes and degraded.
These three challenges—blood stability, T-cell targeting, and efficient endosomal escape—are each world-class challenges in chemistry and biology, and they also hinder one another.Increasing PEG (polyethylene glycol) modification on the LNP surface can prolong circulation time, but excessive PEG hinders cellular uptake and may trigger Accelerated Blood Clearance (ABC) due to anti-PEG antibodies.Increasing the positive charge density of ionizable lipids can improve endosome escape efficiency, but excessively high positive charges can trigger non-specific cytotoxicity, leading to adverse reactions such as injection site pain and complement activation (CARPA).
This is why we say: the core of in vivo CAR-T therapy is not gene editing itself, but the delivery system. CRISPR-edited mRNA can be chemically synthesized, and CAR sequences can be designed to perfection—but if the LNP, this “delivery vehicle,” cannot enter the correct cells, all gene editing technologies are merely beautiful DNA sequences sitting in test tubes, devoid of any clinical significance.
1.2.2 2026 CRS Annual Meeting: A Rational Hub Returning from “Target Frenzy” to “Precision Delivery”
Over the past decade, the entire biopharmaceutical industry has been caught up in a fierce target arms race. Following PD-1/L1 came TIGIT and LAG-3; after TIGIT, ILT3 and VISTA emerged—new targets have been popping up constantly, but only a handful have actually made it through clinical trials.According to statistics from *Nature Reviews Drug Discovery*, over 60% of novel cancer targets that entered clinical development between 2015 and 2025 were ultimately discontinued due to insufficient efficacy or safety concerns. The industry’s collective awakening came slowly but forcefully: the problem wasn’t that the wrong targets were chosen, but that “the drug wasn’t delivered to the right place.”
The annual meeting of the Controlled Release Society (CRS) serves as the premier global forum for DDS researchers. The 2026 conference will take place from July 20 to 23 in Lisbon, Portugal, and is expected to attract over 4,200 attendees from academia, industry, and regulatory agencies.Unlike clinical conferences such as ASCO and ESMO, the core focus of the CRS Annual Meeting has never been “how effective this target is against cancer,” but rather “what material, carrier, or release mechanism have I designed to ensure the drug reaches its intended destination precisely, continuously, and safely.”
The 2026 CRS Annual Meeting holds special historical significance. Over the past 12 months, the field of drug delivery systems (DDS) has been experiencing an unprecedented technological boom.LNP delivery of CAR-T cells in vivo, extrahepatic ligand-conjugation of small nucleic acids, long-acting microspheres and highly absorbable oral formulations for GLP-1 peptides, and smart responsive linkers for ADCs—these four technological pathways achieved landmark breakthroughs almost simultaneously in Q2 2026.They share a common core: all are attempting to solve the fundamental problem of “how to enable drug molecules to breach physiological barriers, precisely reach target tissues, and achieve controlled release.” This is why we say that Lisbon in 2026 will determine the commercial viability of the next generation of cutting-edge biologics—because whoever emerges victorious in this “precision delivery” race will be able to transform non-commercializable biological concepts into mass-producible, profitable clinical products.
| DDS Technology Directions | Representative Pipelines/Companies | Q2 2026 Milestones | Expected Focus at CRS 2026 |
| In vivo CAR-T LNP Delivery | Legend Biotech/BI | First-in-human Phase I data + NHP T-cell targeting validation | Synthesis and liver-bypassing strategies for T-cell-specific LNPs |
| Extrahepatic ligand conjugation of small nucleic acids | Alnylam/Arrowhead | CNS Delivery NHP Data + ANGPTL3 Phase II 18-Month Long-Acting | Coupling Efficiency and Tissue Specificity of Novel Ligand Chemistry |
| Long-acting GLP-1 Microspheres | Novo Nordisk/Eli Lilly | Monthly Microspheres Phase III + Head-to-Head Comparison with Oral Permeation Enhancers | Controlled release of PLGA microspheres + competition with oral peptide carriers |
| ADC Smart Linkers | Daiichi Sankyo/Pfizer/Seagen | Preclinical Validation of Multi-Gate Linkers | Linker Stability + Real-Time Monitoring of Free Toxin Metabolism |
1.3 Overview and Practical Value of This Article
1.3.1 Say Goodbye to Boring Schedules: An Exclusive Breakdown of the Three Major Technological Showdowns in Nucleic Acids, Peptides, and Macromolecules
If you open the official CRS 2026 schedule, you’ll see a dense lineup of over 2,000 posters, hundreds of oral presentations, and dozens of satellite sessions—so much information that you won’t know where to start.What do most industry media outlets do? They dutifully translate the schedule word for word, telling you that “Professor X will deliver a presentation on Topic Y in Room Z,” and then tack on a travel guide to Lisbon—congratulations, you’ve just spent 15 minutes reading a piece of fluff with zero added value.
This article takes a different approach. Starting with the three core topics that attracted the most sponsors, the highest proportion of oral presentation submissions, and the most heated discussions at this year’s CRS Annual Meeting, and combining them with the most cutting-edge global industry trends from Q2 2026, we’ll break down three key technical showdowns for you:First: The Showdown Between Small Nucleic Acids and In Vivo CAR-T for Extrahepatic Targeting via LNP—Which Will Break Through the Liver Barrier First to Deliver Drugs to New Battlefields Like T Cells, the CNS, and Muscle Tissue? Second: The Formulation Race Among GLP-1 Peptides—Long-Acting Microspheres vs. Oral Permeation Enhancers: Which Will Find the Optimal Solution Between “Once a Month” and “One Tablet a Day”?Round 3: The Safety Battle for ADC Linkers—Can multi-gate linkers finally resolve the off-target toxicity that has plagued conjugated drugs for a decade?
In each of these showdowns, we’ll not only explain the underlying principles of the technology but, more importantly, tell you—as an investor, BD professional, or R&D decision-maker—which companies’ posters you should prioritize at the Lisbon conference, how to ask questions that hit the presenter’s pain points, and how to interpret the commercialization roadmap for these technologies over the next 3–5 years. This isn’t a review; it’s a strategic reconnaissance report.
1.3.2 A “Pain Point Focus Guide” for On-Site Attendees and Online Observers
If you’re an R&D professional or BD manager paying out of pocket to attend the conference in Lisbon—with a registration fee of over $5,500, transatlantic flights, and five nights in a hotel—your total investment easily exceeds $10,000.Your time is even more precious. At a conference with 4,200 attendees, every minute should be spent on the most valuable sources of information. This article will tell you directly: which concurrent sessions feature the real heavyweights, which satellite symposia are pure advertisements sponsored by pharmaceutical companies, and which small biotech posters hide the next $1 billion acquisition target.
If you’re an online observer watching webcasts from your office or lab back home: You probably don’t need to worry about which restaurant in Lisbon serves the best paella, but you definitely need to know—can in vivo CAR-T NHP data be translated to humans? How soon will SNAC technology for oral GLP-1 formulations enter clinical trials? Can in vitro data on ADC linker stability predict in vivo toxicity?This article distills the criteria that won’t appear in official press releases—the insights only insiders understand.
2.0 Topic 1 at biotech events: The Clash Between Small Nucleic Acids and In Vivo CAR-T—The Decisive Battle Between Extrahepatic Targeting and Novel LNPs
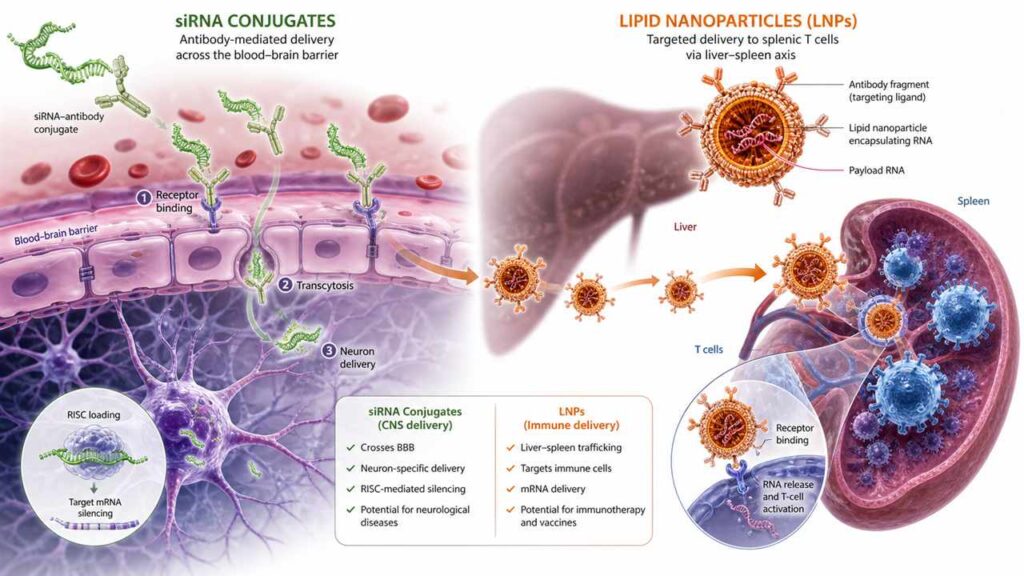
2.1 Focusing on Search Pain Points (SEO Core)
2.1.1 Why Do Existing LNPs Fail to Meet the Therapeutic Needs of In Vivo CAR-T and Autoimmune Diseases?
To answer this question, we must first return to a fundamental anatomical fact: over 90% of any nanoparticles injected intravenously into the human body are captured by the liver.This is not a design flaw, but an evolutionary advantage—the liver’s sinusoidal capillaries possess fenestrations with pore sizes of approximately 100–150 nanometers, which precisely match the size of modern LNPs (typically 50–100 nanometers in diameter).The Kupffer cells (resident macrophages of the liver) and LDL receptors (LDLR) on hepatocytes form a nearly impenetrable hepatic barrier.
For liver diseases (such as liver tumors, hepatitis B, and PCSK9-related familial hypercholesterolemia), the liver’s natural targeting is an advantage. This is why the most successful LNP products to date—Onpattro (for treating hereditary transthyretin amyloidosis) and the two COVID-19 mRNA vaccines—all target the liver as their primary organ.However, when your targets are T cells (CAR-T therapy for lymphoma/SLE), hematopoietic stem cells (gene editing for sickle cell anemia), or neurons (siRNA for Huntington’s disease), the liver becomes a massive “drug black hole”—the vast majority of the dose is absorbed by it, and only a tiny fraction reaches the site where it is truly needed.
Here we introduce a core concept that will be repeatedly mentioned at the CRS Annual Meeting: Liver De-targeting or Extrahepatic Targeting. This is not merely a matter of replacing the LNP’s navigation system.The mechanism by which LNPs are captured by the liver is multi-layered: at the physical level (particle size and surface charge determine hemodynamic distribution), the biochemical level (ApoE in the protein coat determines receptor-mediated hepatic uptake), and the immunological level (phagocytosis and clearance by Kupffer cells). To truly achieve “liver de-targeting,” you need to address all three of these levels simultaneously.
Current major strategies in academia include: First, incorporating anionic or amphiphilic lipids into the LNP formulation to alter the surface charge of the LNP, thereby reducing ApoE adsorption and lowering hepatic uptake.Second, attaching T-cell-targeting ligands—such as anti-CD3, anti-CD5, or anti-CD8 single-chain Fv (scFv) fragments—to the LNP surface, enabling the LNP to actively seek out T cells rather than being passively intercepted by the liver.The third approach utilizes Selective Organ Targeting (SORT) technology. By adjusting the molar ratio of PEG to lipids and the alkyl chain length in the LNP formulation, the biodistribution pattern of the LNP within the body is altered, causing it to accumulate preferentially in the spleen (which is rich in T and B cells) rather than the liver.In 2020, the laboratory of Daniel Siegwart at the University of Texas Southwestern Medical Center first reported the SORT technology in *Nature Nanotechnology*. By 2026, multiple laboratories had validated the ability of SORT-LNP to target splenic T cells in non-human primate (NHP) models.
| Hepatoclave Strategy | Mechanism of Action | Representative Research Groups/Companies | Current Stage | Key Challenges |
| Anionic/Ampholytic Lipids | Reduce ApoE adsorption → Decrease hepatocyte uptake | Acuitas/Moderna mRNA Vaccine LNP | Phase III clinical trials (liver-targeting maturation) | T-cell specificity remains insufficient |
| T-cell-targeted ligand conjugation (scFv) | Anti-CD3/CD5/CD8 antibodies guide LNP → T cells | Legend Biotech/BI/Ovana Bio | Phase I clinical trial (first-in-human data) | Ligand stability + immunogenicity |
| SORT (Selective Organ Targeting) | Adjusting PEG-lipid ratio to alter biodistribution | Siegwart Lab/ReCode Therapeutics | NHP validation completed | Formulation optimization requires large-scale screening |
| Biodegradable PEG-lipid | pH-sensitive PEG detaches within endosomes → Enhanced escape | Multiple academic laboratories | Preclinical (mice/non-human primates) | Lack of long-term safety data |
2.1.2 Recent Advances in Materials Science to Overcome “Liver Uptake”
If 2020–2024 was the golden age of CRISPR gene editing tool evolution, then 2024–2026 will be the renaissance of lipid chemistry. The synthesis of novel ionizable cationic lipids has moved beyond a phase of “trial and error” and entered a new era of “rational design + AI-assisted screening.”
The design logic of traditional ionizable cationic lipids (such as SM-102 and ALC-0315 used in the Moderna and Pfizer/BioNTech vaccines) is relatively simple: an ionizable head group (typically a tertiary amine) is linked via a connecting arm to one or more hydrophobic alkyl tail chains.At acidic pH, the tertiary amine becomes protonated and carries a positive charge, helping the LNP encapsulate the negatively charged mRNA and interact electrostatically with the endosomal membrane.At physiological pH, the tertiary amine deprotonates and returns to a neutral state, reducing cytotoxicity. This pH-responsive charge-flipping mechanism is the cornerstone of LNP technology. However, it is far from sufficient to solve the problem of liver accumulation—traditional SM-102-type lipids are almost inevitably captured by the liver’s ApoE-LDLR pathway.
Breakthroughs in 2025–2026 will stem from two innovative approaches.The first is the design of “biodegradable lipids.” By introducing chemical groups—such as ester bonds, disulfide bonds, or acetal bonds—that can be cleaved by enzymatic hydrolysis in vivo into the linker arms or alkyl tail chains of lipid molecules, LNPs can rapidly degrade after completing their task, thereby reducing chronic toxicity caused by long-term accumulation.The laboratories of Robert Langer at MIT and James Dahlman at Georgia Tech have taken the lead in this area—in a paper published in late 2025, they demonstrated a set of ultra-rapidly degrading lipids incorporating multiple ester bonds, reducing the half-life in mouse livers from several weeks for traditional LNPs to less than 48 hours, while mRNA delivery efficiency not only remained unchanged but actually increased by 30% due to the rapid unlocking mechanism.
The second approach is even more ambitious: large-scale combinatorial screening of the “Extrahepatic Lipid Library.” The Dahlman lab established a library containing thousands of lipid molecules with diverse chemical structures. Using DNA barcoding technology, they tagged each LNP with a unique DNA sequence and injected them all at once into mice.Twenty-four hours later, by analyzing the abundance of DNA barcodes in various tissues via high-throughput sequencing, they could deduce which lipid structures naturally prefer which organs. This method is known as FIND (Fast Identification of Nanoparticle Delivery) technology.Using this method, they identified several classes of atypical lipids that preferentially target the spleen, bone marrow, and even the lungs. These lipids share common features, such as branched alkyl tail chains and irregular molecular geometries, which may enable selective binding to specific receptors on the surface of vascular endothelial cells in these organs.
These findings are being extensively reported at CRS 2026. According to abstracts leaked prior to the conference, at least two independent research groups will present entirely new spleen-targeting lipids identified using FIND or similar technologies, backed by NHP data. If these NHP data replicate the high transduction efficiency observed in mice, the loop for in vivo CAR-T delivery will be truly closed.
2.2 Predictions for Key Debates at CRS 2026
2.2.1 The Evolution of Novel Cationic Lipid Synthesis: How to Balance Endosomal Escape Efficiency and Immunogenicity?
At CRS 2026, the number of oral presentations and posters on novel cationic lipids is expected to reach an all-time high. The central debate can be summarized as a dilemma: the trade-off between endosome escape efficiency and immunogenicity.
To understand this dilemma, we must revisit the fundamental physicochemistry of LNP delivery. After being taken up by cells, LNPs become trapped within endosomes. The internal environment of endosomes is slightly acidic (pH 5.0–6.5), and this acidic environment triggers the protonation of ionizable cationic lipids.The protonated cationic lipids interact electrostatically with anionic phospholipids (such as phosphatidylserine) on the endosomal membrane, inducing the endosomal membrane lipids to transition from a bilayer arrangement to a hexagonal HII phase. This creates pores in the membrane or completely disrupts its integrity, allowing the mRNA to escape into the cytoplasm.The efficiency of this process depends on two key parameters: the lipid’s pKa value (the pH threshold required for protonation) and the lipid molecule’s ability to induce a non-bilayer phase in the membrane (a parameter related to the conical shape of the molecule).
Theoretically, the higher the pKa value (the easier it is to protonate) and the more pronounced the molecular conicity (the easier it is to disrupt the membrane structure), the higher the efficiency of endosome escape. However, the problem is that cationic lipids with excessively high pKa values will partially protonate at blood pH (7.4), creating a positively charged surface—which leads to two catastrophic consequences:First, positively charged LNPs will non-specifically adsorb plasma proteins, altering the composition of the protein coat and exacerbating hepatic capture; second, the positively charged surface will activate the complement system (complement-associated pseudo-anaphylactic reaction, CARPA), leading to acute inflammatory responses, thrombocytopenia, and even anaphylactic shock.
Is there a lipid molecule that is completely uncharged (low immunogenicity) at pH 7.4, fully protonated (high escape efficiency) at pH 6.0, and forms a non-bilayer phase with high reversibility (avoiding permanent cell membrane disruption and toxicity)? This is the Holy Grail of cationic lipid chemistry in 2026. According to pre-conference abstracts,the Anderson Lab at MIT and the Mitchell Lab at the University of Pennsylvania will present their independently developed next-generation ionizable lipids at CRS 2026—both with pKa values precisely controlled between 6.2 and 6.4, completely neutral at physiological pH, protonation rates exceeding 95% at endosomal pH, and no CARPA reactions observed following either intramuscular or intravenous administration.If these data hold up, the fundamental chemical components for CAR-T LNPs in vivo will essentially be complete.
| Lipid Design Parameters | Traditional Design (SM-102, etc.) | Next-generation design (expected for CRS 2026) | Significance for In Vivo CAR-T |
| pKa value | 6.5–6.9 (relatively broad) | 6.2–6.4 (precise and narrow) | Reduces non-specific positive charge at physiological pH |
| Molecular taper parameters | Moderate | Highly tapered (enhanced membrane disruption) | Increases endosome escape efficiency to 5–10% |
| Biodegradability | Limited (partial ester bonds) | Multiple ester bonds/disulfide bonds/acetal bonds | Reduces tissue accumulation and chronic toxicity |
| Immunogenicity | CARPA risk in some batches | NHP-validated as CARPA-free | Widening the safety dose window |
| Liver targeting | Strong (natural ApoE binding) | Can be regulated via formulation (SORT) | Delivery to splenic T cells is feasible |
2.2.2 Industrial Implementation and Safety Data for Organ-Specific Delivery (e.g., SORT Technology)
SORT (Selective Organ Targeting) is one of the most exciting conceptual breakthroughs in the field of drug delivery systems (DDS) since 2020.Its core discovery is surprisingly simple: by adjusting the molar ratio of PEG-lipids and the alkyl tail length in the LNP formulation, one can systematically alter the biodistribution of LNPs in vivo—shifting from almost complete accumulation in the liver to preferential accumulation in the spleen, lungs, or even bone marrow.
What is the mechanism behind this? The current leading hypothesis is that different PEG-lipid combinations alter the composition of the protein corona formed on the LNP surface.For example, LNPs prepared with short-chain PEG-lipids (C14) tend to adsorb more vitronectin and fibrinogen; these proteins interact with integrin receptors in the spleen and lungs, directing the LNPs to these organs.In contrast, LNPs prepared with long-chain PEG-lipids (C18) preferentially bind to ApoE, maintaining liver targeting. This mechanism was experimentally validated through proteomic analysis in 2025.
However, the industrial implementation of SORT technology faces a unique challenge: reproducibility. The SORT effect is highly dependent on the precise ratio of the LNP formulation—a 1–2 percentage point change in the molar ratio of PEG-lipids can cause the organ distribution pattern to shift from spleen preference to liver preference.While this level of precision is achievable in small-scale, milligram-level laboratory preparations, maintaining such exact formulation ratios in industrial-scale production—ranging from hundreds of grams to kilograms—presents a significant CMC (Chemistry, Manufacturing, and Control) challenge. This explains why, despite the SORT technology having been published six years ago, the first validation data for SORT-LNPs in NHPs did not emerge until 2026—as NHP studies require sufficiently large and stable LNP batches.
At CRS 2026, oral presentations are expected from ReCode Therapeutics and several Asian biotech companies, focusing on the biodistribution and preliminary safety data of SORT-LNP in NHPs. If these data demonstrate that the SORT effect is reproducible in NHPs and that a clear dose-response relationship exists, then the final mile in the journey of SORT technology—from academic concept to industrial application—will have been completed.
2.3 Commentary from a U.S. Industry Perspective (AI-Filtered)
2.3.1 The Capital Market’s Ruthless Shift: Focusing Exclusively on Real-World Data from Non-Human Primates (NHPs)
The capital markets of 2026 will no longer buy into flashy PowerPoint presentations based on mouse experiments—this may sound simple, but only those who have truly weathered the biotech investment and financing winter of the past 18 months know just how brutal it is.If the bubble era of 2020–2021—when “any company with the letters LNP in its name could raise $50 million”—is truly gone for good, then the funding environment in Q2 2026 is an absolute buyer’s market. Investors on Wall Street and Sand Hill Road now ask only three questions: First, where are your NHP data? Second, what is the absolute value of your extrahepatic transfection efficiency?Third, can your LNP batches be scaled up to clinical-grade at the CDMO? If you can’t answer any one of these three questions, this funding round is over.
The direct consequence of this shift is that the “quality” of companies at CRS 2026 will be sharply polarized.Investors will walk right past the booths of companies that only display mouse bioluminescence imaging (IVIS) images on posters, without even giving them a second glance. Meanwhile, companies that can present NHP PET/CT images during oral presentations and demonstrate the absolute copy number of CAR mRNA in splenic T cells will be surrounded by crowds of people.
For those of us in the audience, the quick rule of thumb for determining whether an LNP biotech company is worth a deeper conversation is simple: does its data table include a row for NHPs? If so, it’s worth a 15-minute chat. If not, treat it as wallpaper on the poster board.
2.3.2 Guide to Attending and Observing: Keep a Close Eye on Biotech Booths Presenting NHP Data
Specifically for the CRS 2026 Exhibition Hall, I recommend allocating your time as follows: On the morning of the first day, spend an hour quickly scanning the entire exhibition area, using your phone to photograph the company names and poster numbers at every booth.Then return to your hotel and use the pre-conference abstracts and the companies’ pipeline information on ClinicalTrials.gov to categorize them into three tiers: Tier A (in vivo CAR-T/small-nucleic acid LNP companies that have published NHP data), Tier B (companies with NHP data but for non-core indications), and Tier C (companies with only mouse data or those in the purely conceptual stage).On the second and third days, spend 70% of your time at Category A booths and 20% at Category B booths. Unless you’re seeking academic collaborations, don’t waste time on Category C booths.
At this 4,200-person conference, information overload is your biggest enemy.Bring a sheet of A4 paper listing the three technical questions you most want to verify (e.g., “What is the absolute transduction efficiency of your LNP in NHP splenic T cells?” or “What percentage of the total ADC is the free payload’s AUC in plasma?”). Fill in one box after each conversation with a company—by the end of the three days, this A4 sheet will be your most valuable investment report of the year.
3.0 Topic 2 at biotech events: The “Endgame of Hypercompetition” Between Peptides and GLP-1—The Battle Between “Once-Monthly” Microsphere Technology and Highly Absorbable Oral Formulations
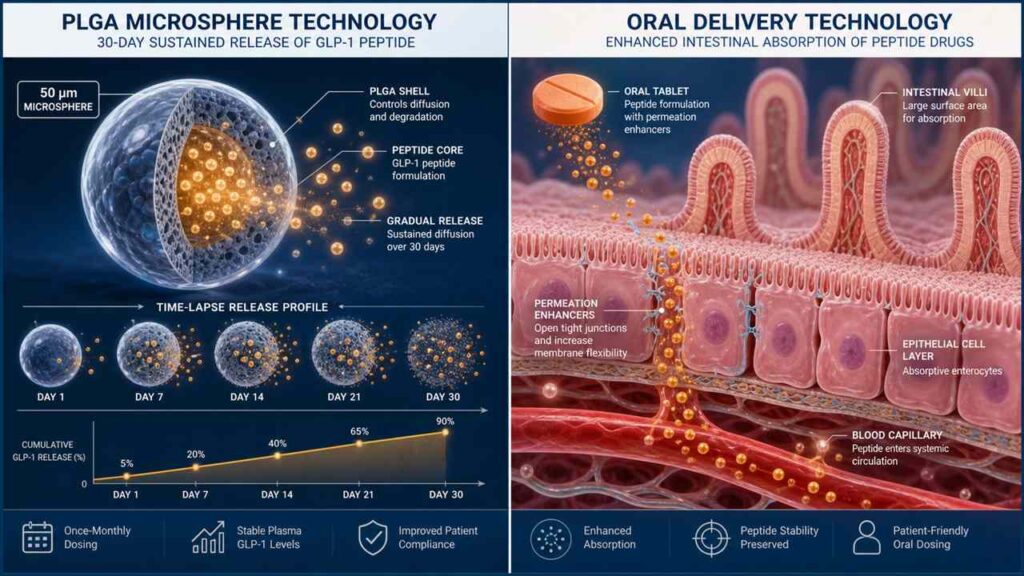
3.1 Industry Landscape and Intensity of Commercial Competition
3.1.1 The Latest Pipeline Showdown Between Novo Nordisk and Eli Lilly in Q2 and the Expansion into Multiple Indications
If 2023 was the year GLP-1 drugs broke into the mainstream—the year semaglutide (brand names Ozempic/Wegovy) and tirzepatide(Tirzepatide, brand names Mounjaro/Zepbound) transitioned from diabetes medications to global consumer brands—then 2024–2026 will be the years of an all-out arms race in the GLP-1 sector. Global sales are projected to exceed $80 billion in 2025, with analysts widely predicting they will surpass $150 billion by 2030. This market is so vast that no multinational pharmaceutical company can afford to sit idly by.
In Q2 2026, Novo Nordisk and Eli Lilly simultaneously released three major announcements capable of reshaping the competitive landscape. First, Novo Nordisk published Phase IIIb data for high-dose versions of oral semaglutide (25mg and 50mg).The previously approved oral semaglutide had a maximum dose of 14mg (brand name Rybelsus), but due to the absorption efficiency ceiling of the SNAC permeation enhancer, its overall bioavailability was a meager 0.5%–1%. The high-dose versions aim to trade quantity for efficacy—increasing the dosage to compensate for the low absorption rate.The results were indeed promising: the 50mg dose group achieved glycemic control and weight loss effects comparable to those of 2.0mg semaglutide injections. However, this came with a downside: the incidence of gastrointestinal side effects (nausea, diarrhea, vomiting) caused by high-dose SNAC surged to nearly 40%, posing a severe challenge to patient adherence.
Second, Eli Lilly released the first Phase II data for its next-generation triple agonist (GIP/GLP-1/Glucagon, code-named Retatrutide). The results showed that after 48 weeks of treatment, liver fat content decreased by an average of over 80%, and the rate of fibrosis improvement was significantly better than that of the placebo.The significance of these data lies in the fact that, as competition in the weight-loss market between GLP-1 monotherapy and GIP/GLP-1 dual-target therapies intensifies, Eli Lilly is quietly expanding its focus from weight loss to NASH (non-alcoholic steatohepatitis)—a blue ocean where competition is still relatively limited.
Third, and most relevant to CRS 2026—both Novo Nordisk and Eli Lilly released a flurry of updates in Q2 regarding the latest R&D progress on “once-monthly” long-acting injectable formulations.Novo Nordisk’s strategy relies on osmotic pump technology, involving the subcutaneous implantation of a rice-grain-sized micro-osmotic pump to enable slow drug release; Eli Lilly’s strategy, however, utilizes traditional PLGA microsphere technology, encapsulating GLP-1 peptides within biodegradable polymer microspheres, with the peptides released through the gradual hydrolysis of the PLGA within the body.Phase III data for both strategies indicate that the “once-monthly” goal, which once seemed unattainable, is now nearing technical maturity. However, what truly determines the winner has never been whether the product can be developed, but rather whether the costs can be sustained and side effects effectively managed after commercialization.
| Competitive Landscape | Novo Nordisk | Eli Lilly | Key Milestones in Q2 2026 |
| Flagship Products | Semaglutide (injection + oral) | Tirpepetide (GIP/GLP-1 dual-target injection) | Launch of high-dose oral formulation; Retatrutide MASH data |
| Long-acting strategy | Subcutaneous Implantation of Osmotic Pumps | PLGA microspheres encapsulating peptides | Both have entered Phase III clinical trials |
| Oral route | SNAC Osmolytic Enhancer → High-Dose Breakthrough | Next-generation transcellular permeation enhancer | Novo Nordisk 25/50 mg approved; Eli Lilly in Phase I of oral formulation |
| New indications | MASH/CVD/Alzheimer’s disease | MASH/Sleep Apnea/Heart Failure | Retatrutide Delivers Impressive Phase II Data for NASH |
3.1.2 Compliance Economics: Comparing the Commercial Value of Long-Acting Injectables and Convenient Oral Formulations
In the GLP-1 therapeutic landscape, there is an underappreciated core concept: “Compliance Economics.” Simply put, the actual efficacy of a drug equals its theoretical efficacy multiplied by the frequency at which patients actually take it.If a drug theoretically has an efficacy of 100 points, but patients actually take only 50% of the prescribed dose (the six-month discontinuation rate for GLP-1 in the real world is approximately 40%–60%), then its actual clinical value is only 50 points.
This is why the dosage form determines the commercial success or failure of GLP-1 drugs. Currently, the best-selling GLP-1 product on the market is semaglutide, which is administered once a week—which sounds convenient enough. However, real-world data shows that even with this extremely low-frequency dosing regimen, nearly one-third of patients discontinue treatment within six months for various reasons.Reasons include, but are not limited to: fear of needles (needle phobia), injection site reactions (redness, swelling, hard lumps), forgetting to bring medication while traveling or on business trips, or simply finding the act of injecting at a fixed time each week to be a psychological burden.
So what about the “once-a-month” microsphere/osmotic pump injection? It can significantly reduce the window for forgetting to take the injection—lowering the pressure from once a week to once a month, which represents a qualitative leap in the psychological barrier for many patients.But what is the trade-off? The trade-off is an initial burst release—in the first few days after injection, the microspheres may release far more than the safe dose of the peptide, causing patients to experience severe nausea and vomiting. This is why, in the Phase III data for the “once-monthly” formulations from Eli Lilly and Novo Nordisk, the incidence of gastrointestinal adverse events in the first two weeks was significantly higher than that of the weekly formulations.
On the other hand, an oral GLP-1 tablet that is “as convenient as taking cold medicine” is undoubtedly the ultimate dream. The problem, however, is that the oral bioavailability of peptides is inherently very low (due to high molecular weight, susceptibility to degradation by stomach acid, and poor permeability through the intestinal epithelium).The only currently successful oral GLP-1—semaglutide tablets—relies on SNAC (sodium 8-(2-hydroxybenzamido)octanoate), a permeability enhancer.SNAC creates a small local window in the stomach by temporarily increasing the transmembrane permeability of gastric mucosal epithelial cells, allowing the peptide molecules to squeeze through. However, the side effects of this mechanism are also significant—it requires a certain degree of irritation to the gastric mucosa. This is why oral semaglutide must be taken strictly on an empty stomach, and a significant proportion of patients experience nausea and stomach discomfort.
The key focus of CRS 2026 is: Will a second-generation permeation enhancer emerge that is gentler and more effective than SNAC? Are there new intestinal targeted-release technologies capable of safely delivering the peptide to the small intestine—where absorption is highest—rather than causing mucosal damage in the stomach? These are the challenges formulation engineers are currently grappling with.
3.2 CRS 2026 Sponsors and Key Academic Highlights
3.2.1 High-Loading, Long-Acting Microsphere Technology: How to Overcome the “Initial Burst Release” of PLGA Microspheres and Peptide Degradation?
PLGA (poly(lactic-co-glycolic acid)) is the most classic and widely used biodegradable polymer in the field of sustained-release drug delivery.It has been FDA-approved for human use for over 30 years, and from leuprolide to risperidone microspheres, PLGA has proven its safety and degradability. However, when it comes to peptide drugs like GLP-1, two long-standing issues resurface: initial burst release and peptide degradation within the microspheres.
How does early burst release occur? When PLGA microspheres are injected into subcutaneous or intramuscular tissue, the peptide molecules adsorbed on or near the microsphere surface dissolve immediately and diffuse into the bloodstream, creating a concentration spike at the onset of administration.This peak can reach 5–10 times the steady-state plasma concentration—for peptides like GLP-1, which already have significant gastrointestinal side effects, this is the direct cause of patients vomiting so severely after injection that they question their very existence. Moreover, the rapid release is immediately followed by a trough—since a large amount of the drug is released prematurely, the subsequent sustained-release phase fails to maintain therapeutic efficacy.
Peptide degradation within the microspheres is another major challenge. PLGA degrades in vivo through hydrolysis—its ester bonds break under the attack of water molecules, producing lactic acid and hydroxyacetic acid.However, this process itself creates an acidic microenvironment inside the microspheres (pH can drop below 3.0), and peptides are extremely unstable in acidic environments, prone to deamidation, oxidation, and aggregation, leading to loss of drug activity. The combined effect of these two issues is that of the injected GLP-1 peptide, only 60%–80% may ultimately be released into the bloodstream in its intact, active form.
A series of new solutions addressing these two issues will be presented at CRS 2026. One of the most promising approaches is the “core-shell microsphere” structure: by encapsulating the peptide drug within a core composed of PLGA with different molecular weights or varying lactic acid/glycolic acid ratios, and then coating it with a more hydrophobic shell, zero-order release of the peptide is achieved—constant, sustained, and free of sudden spikes.Another cutting-edge approach involves introducing basic additives (such as zinc carbonate or magnesium oxide) into the microsphere matrix. By leveraging their buffering capacity to neutralize the acidic byproducts of PLGA hydrolysis, these additives protect the chemical stability of the peptides.Another team is working on covalent drug-PLGA conjugation—not physical encapsulation, but directly linking the peptide to the PLGA polymer chain via hydrolyzable chemical bonds. The peptide is released only when the PLGA hydrolyzes and breaks, fundamentally eliminating peak release.
| Challenges with PLGA Microspheres | Traditional Methods | New Solution at CRS 2026 | R&D Challenges |
| Initial Release Peak | Rapid Drug Release via Surface Adsorption | Core-shell structure + dense surface coating/secondary coating | Increased process complexity → Difficulties in industrial scale-up |
| Peptide degradation within the microspheres | PLGA hydrolysis creates an acidic microenvironment (pH < 3) | Alkaline additives (ZnCO₃/MgO) buffer the pH | Long-term safety of additives must be verified |
| Non-uniform release | Uneven PLGA molecular weight distribution → Heterogeneous degradation | Monodisperse PLGA + precise molecular weight control | The synthesis cost of monodisperse PLGA is extremely high |
| Insufficient drug loading | Traditional W/O/W emulsification method: encapsulation efficiency 30–50% | Microfluidic precise spheronization + covalent conjugation | Engineering bottlenecks in high-throughput microfluidic production |
3.2.2 Next-Generation Permeation Enhancers for Oral Peptides: New Carrier Competitors Transcending the SNAC Mechanism
As the first successfully commercialized oral peptide permeability enhancer, SNAC deserves to be recognized as a historic breakthrough—it transformed peptides from a treatment that required injections into one that could be taken orally.However, the industry has also fully recognized SNAC’s limitations: bioavailability remains extremely low (0.5%–1%), meaning over 99% of the drug is wasted; efficacy is highly dependent on strict fasting conditions, with absorption rates potentially dropping by more than 50% when taken after meals; and gastrointestinal tolerability remains an issue.
At CRS 2026, oral peptide delivery will be one of the hottest topics.The new generation of technological approaches can be broadly divided into several categories. The first is the gut-targeted release system: by coating the tablet surface with a pH-responsive enteric coating, the tablet passes intact through the stomach (pH 1–2) and only disintegrates to release the peptide and permeation enhancers upon reaching the small intestine (pH 6–7), thereby avoiding the gastric irritation caused by SNAC’s direct interaction with the gastric mucosa.The second approach is an upgraded version of transcellular permeation enhancers—shifting from SNAC’s paracellular pathway to a transcellular pathway. This method actively mediates the passage of peptide molecules through the intestinal epithelial cells themselves for absorption, without disrupting the intercellular barrier. Eli Lilly and several biotech companies are currently developing natural transcellular transport mechanisms based on bile acid transporters (ASBT) or vitamin B12 receptors.
The third approach is the self-microemulsifying drug delivery system (SMEDDS)—dissolving the peptide in a mixture of oil, surfactants, and co-surfactants. Upon oral administration, this mixture spontaneously forms extremely fine microemulsion droplets (particle size <100 nm) when exposed to the aqueous environment of the gastrointestinal tract. These droplets are absorbed via the intestinal lymphatic system, directly bypassing the first-pass effect of the liver.The fourth, and most innovative, approach involves oral microneedle capsules. The core concept of this technology is not to attempt to allow peptide molecules to permeate the intestinal barrier, but rather to physically breach it: the patient swallows a capsule containing a soluble microneedle array; the capsule unfolds within the intestine; the microneedles painlessly penetrate the intestinal mucosa to inject the drug into the intestinal wall; and the microneedles then dissolve on their own.The Langer and Traverso teams at MIT have already validated the concept of microneedle capsule delivery of insulin in pig models and received FDA IND approval by the end of 2025.
3.3 Differentiated In-Depth Insights
3.3.1 Looking Beyond Clinical Data to the Essence: The “Scale-Up Effect” of Formulation Processes and Commercial Cost Control
Data on long-acting GLP-1 microspheres presented at academic conferences always look impressive—immediate release rate <5%, peptide activity retention >95%, and sustained release for 30 days—but these data almost exclusively come from small-scale laboratory trials ranging from milligram to gram levels.The real test has never been whether the formulation can be produced, but whether the same quality can be maintained after scaling up to the hundred-kilogram level. This concept, known in the pharmaceutical industry as the “scale-up effect,” is a pipeline killer for many biotech companies.
Why is the commercial scale-up of PLGA microspheres so difficult? Because when preparing microspheres in the lab, you typically use solvent evaporation or microfluidic technology—precisely controlling stirring speed, temperature, and solution concentration—resulting in microspheres with highly uniform particle size (coefficient of variation CV < 10%).However, when scaling up to industrial-scale reactors (300–1,000 liters), factors such as uneven mixing, local temperature gradients, and limitations in mass and heat transfer can cause the microsphere size distribution to widen—with some microspheres as small as 20 micrometers and others expanding to 200 micrometers.Inconsistent particle size leads to inconsistent release rates—larger microspheres release slowly, while smaller ones release quickly, making the drug release profile of the entire batch unpredictable. This is why the FDA imposes extremely stringent requirements for intra-batch and inter-batch consistency of microsphere products, and why only a handful of companies worldwide have successfully commercialized PLGA microsphere products.
At the same time, cost issues are even more acute. GLP-1 peptides themselves are already exorbitantly expensive at the amino acid level—chemically synthesizing a linear peptide with more than 30 amino acids costs anywhere from several hundred to several thousand dollars per gram.Coupled with yield losses from the PLGA microsphere encapsulation process (typically resulting in a 20%–40% loss of the peptide during encapsulation), the final cost per injection could reach as high as $1,000–$3,000. If payers refuse to pay a higher premium for the marginal convenience of “once-monthly” versus “once-weekly” dosing, this long-acting formulation could be eliminated during the competitive bidding process.
| Scaling Challenges | Pilot Scale (1–100 g) | Pilot Scale (1–10 kg) | Commercial use (≥100 kg) | Countermeasures |
| Microsphere Particle Size CV% | <10% (microfluidics) | 15–25% | >30% (industrial reactor) | Online Particle Size Monitoring + PAT Process Analytical Technology |
| Peptide activity retention | >95% | 85–92% | 70–85% (acidic degradation) | Alkaline additives + low-temperature freeze-drying |
| Batch-to-batch consistency | Good | Moderate | Extremely challenging | QbD (Quality by Design) + DoE |
| Per-unit cost | $200–500 | $500–1,000 | $1,000–3,000 | Process Optimization + Automation + Economies of Scale |
3.3.2 Geographical Differences in Formulation Design Among European and American Pharmaceutical Companies
If you pay close attention to the nationality distribution of exhibitors at CRS 2026, you will notice an interesting phenomenon: The booths of U.S. companies are dominated by posters and prototype samples of various oral permeation enhancers, transcellular transport carriers, and gut-targeted release tablets;whereas the booths of European and Japanese companies are almost exclusively filled with process demonstrations of complex long-acting microspheres, osmotic pump implants, and precision injection devices. This is no coincidence—it reflects a deep-seated difference in geographically-driven technological thinking.
The core strategy of U.S. pharmaceutical companies in GLP-1 formulation innovation is to “go all-in on oral delivery.” This choice is driven by profound commercial logic: the U.S. is the world’s largest GLP-1 consumer market, and the market’s core driver is not the rigor of clinical use, but consumers’ demand for convenience.Under the U.S. DTC and commercial insurance systems, if a drug can be formulated as an oral tablet that is “as simple to take as cold medicine,” it can reach tens of millions of patients who have a “psychological aversion to injections” but are “willing to take pills.”This is why Eli Lilly views oral GLP-1 as its next billion-dollar growth driver—not because oral tablets are more effective than injections, but because they can reach new users that injections cannot.
To achieve this goal, the U.S. drug formulation innovation strategy is primarily focused on two directions.First, relentless pursuit of chemical modifications. Eli Lilly and several biotech companies are developing various GLP-1 analogs modified with non-natural amino acids (such as Amino Acid Substituted Analogs). By introducing non-natural amino acids (such as Aib or N-Me-Ala) at key sites in the peptide, these analogs resist degradation by intestinal proteases while maintaining activity at the GLP-1 receptor.This type of chemical innovation is a traditional strength of U.S. pharmaceutical companies—historically, semaglutide itself achieved resistance to intestinal proteases by introducing Aib at the eighth position of the peptide, and this chemical modification is also one of the prerequisites for its absorption to be enhanced by SNACs. Second, innovation in the development of permeability enhancers.SNAC represents the first generation; within the U.S. innovation ecosystem, second- and third-generation permeation enhancers are emerging—ranging from transmembrane enhancers based on bile acid transporters, to active transport carriers based on vitamin B12 receptors, to gut-targeted adhesion systems based on nanopowders.The common thread among these technological approaches is that they all attempt to use chemical or biological methods to “trick” the intestinal barrier, allowing peptide molecules to pass through.
In contrast, the approach of European and Japanese pharmaceutical companies is entirely different. Their core strategy can be summarized as “long-acting injections achieved through engineering ingenuity.” Novo Nordisk’s subcutaneous osmotic pump implant—a rice-grain-sized microdevice that slowly releases medication for 30–90 days after subcutaneous implantation—is a quintessential product of European engineering thinking.Japan’s leuprolide microspheres—the world’s first successfully commercialized PLGA sustained-release microsphere product—equally exemplify the East Asian pharmaceutical industry’s relentless pursuit of complex manufacturing processes.The root of this difference lies in the fact that healthcare systems in Europe and Japan rely more heavily on physicians’ prescriptions and injection procedures, and patients there are significantly more accepting of “going to the hospital for a monthly injection” than their American counterparts (in Germany and Japan, the primary settings for chronic disease management are general practitioners’ offices and specialist hospitals, where injections are routine procedures);simultaneously, the insurance reimbursement systems in Europe and Japan prioritize “duration of therapeutic effect” over “convenience of administration”—if a monthly injection ensures stable blood drug concentrations and sustained efficacy, insurers are willing to pay for this “certainty.”
From a technical perspective, these two differing mindsets present distinct engineering challenges.The challenge with the U.S. approach lies in the “curse of low bioavailability”—when bioavailability is only 0.5%, even a minor fluctuation in absorption rate is amplified 200-fold, leading to significant variability in clinical efficacy. Furthermore, it is extremely difficult to completely eliminate the food effect in oral formulations—even with second-generation permeation enhancers, it is hard to ensure that absorption rates for a patient who has just eaten a hearty breakfast and a patient on an empty stomach remain in the same order of magnitude.The challenge with the European and Japanese approach lies in “process complexity”—the manufacturing processes for PLGA microspheres and osmotic pumps are extremely intricate. From temperature control during emulsification and solvent evaporation to particle size distribution analysis of the microspheres, and from the assembly of the osmotic pump’s semipermeable membrane to aseptic filling, every step is a rigorous CMC test.However, once these manufacturing hurdles are overcome, the technical barriers for long-acting injectable formulations become correspondingly higher—competitors will find it difficult to catch up, as this requires years of accumulated process expertise rather than merely capital investment.
For investors and business development professionals, the practical lesson from this geographical mindset difference is this: if you’re evaluating a U.S. biotech company developing an oral GLP-1 drug, don’t just look at its mouse bioavailability data; focus instead on its food effect studies and clinical consistency data—because food effects are the Achilles’ heel of oral peptides.If you’re evaluating a European or Japanese company developing long-acting microspheres, don’t just look at its small-scale trial data; instead, focus on its pilot-scale and scaled-up commercial data—because successful scaling is the make-or-break factor for microsphere products. Ultimately, these two technological pathways may not result in one replacing the other, but rather in a tiered coexistence: oral formulations will dominate the consumer-grade market for mild to moderate weight loss and metabolic health, while long-acting injectables will dominate the medical-grade market for severe obesity and diabetes.
| Dimensions | The U.S. Approach (Pushing Hard on Oral Formulations) | European/Japanese Approach (Mastering Long-Acting Injectable Formulations) |
| Core Drivers | Consumer Convenience / Acquiring New Users | Clinical Certainty / Sustained Efficacy |
| Representative Technologies | Chemically Modified Peptides + Enhanced Permeation Promoters | PLGA Microspheres/Osmotic Pump Implants |
| Core Engineering Challenges | Low Bioavailability Scale-up Variability + Food Effects | Process Scale-Up + Batch-to-Batch Consistency |
| Technical Barriers | Low (chemical innovations can be rapidly adopted) | High (difficult to replicate accumulated process expertise) |
| Insurance reimbursement rationale | Convenience Premium — Reaching New Patients | Certainty premium — more stable efficacy |
| Dominant Market | Mild to moderate weight loss/metabolic health (consumer-grade) | Severe Obesity/Diabetes: Medical-Grade |
4.0 Topic 3 at biotech events: The Safety Ceiling of ADCs—Timed and Site-Specific Cleavage of Linkers and In Vivo Circulatory Stability
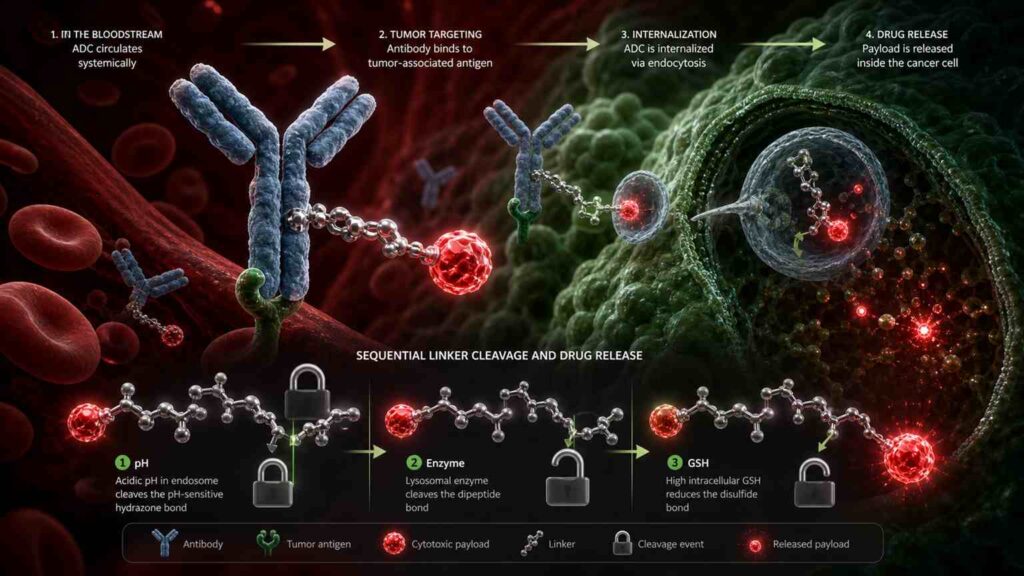
4.1 Core Challenge: The Looming Threat of “Off-Target Toxicity” Behind the “All-Round Efficacy” of ADCs
4.1.1 Severe Side Effects Caused by Premature Release of Macromolecular Conjugates (XDCs) During In Vivo Circulation
Antibody-drug conjugates (ADCs) are hailed as biological missiles for cancer treatment.Their design logic sounds flawless: using antibodies (a precision guidance system) to deliver chemotherapy toxins (warheads) precisely to the surface of cancer cells; after antigen-antibody-specific binding, the ADC is internalized by the cancer cell; the linker is cleaved intracellularly, releasing the toxin to kill the cancer cell—while healthy cells remain completely unaffected because they do not express the target antigen.If this mechanism truly functioned 100% as designed, ADCs would be the most perfect form of chemotherapy ever developed. However, the reality is that ADCs’ clinical performance falls far short of this ideal.
The problem lies in off-target toxicity—before the ADC reaches the cancer cells, the linker breaks prematurely in the bloodstream, releasing the free chemotherapy toxin into the blood, which indiscriminately attacks rapidly dividing cells such as bone marrow hematopoietic cells, lung epithelial cells, and nerve cells.As a result, although patients are receiving precision-targeted ADCs, they suffer from side effects nearly identical to those of conventional chemotherapy: severe neutropenia, thrombocytopenia, interstitial lung disease (ILD), and peripheral neuropathy.
There are two primary mechanisms for premature linker cleavage. The first is chemical instability—if the chemical bonds used in the linker have a half-life that is too short at physiological pH (7.4), spontaneous hydrolysis will occur during circulation.The SMCC sulfide linker used in the classic drug Kadcyla (T-DM1, trastuzumab emtansine) is claimed to be unbreakable, but in reality, the rate of free DM1 release in circulation is approximately 1%–2% per day. Cumulatively, a significant proportion of the toxin is released prematurely within the 2–3 week dosing interval.The second mechanism is enzymatic instability—esterases and proteases in the blood may non-specifically cleave the linker. This is particularly common in early linker designs based on ester bonds.
By 2026, the ADC field had transitioned from the question of “whether ADCs can be developed” to a new phase focused on “whether the developed ADCs are safe.” A landmark event occurred between late 2025 and Q2 2026, when at least two ADC pipelines in Phase II/III clinical trials were partially suspended by the FDA due to data on severe interstitial lung disease (ILD).In one of these cases, the incidence of ILD reached as high as 15%, with Grade 3 or higher ILD occurring in 4% of patients—far exceeding the tolerance thresholds set by regulators and clinicians. Postmortem analysis revealed that the concentration of free toxin detected in ILD-affected lung tissue was several times higher than that in tumor tissue—directly indicating that existing linker technologies are still far from sufficient to support the truly safe use of ADCs.
| ADC Off-Target Toxicity | Common Manifestations | Root Causes | Incidence (representative of ADCs) | Clinical Consequences |
| Hematologic Toxicity | Neutropenia/Thrombocytopenia | Free payload attacks bone marrow hematopoietic cells | 30–70% (varying by ADC) | Dose-limiting toxicity → Treatment interruption |
| Interstitial lung disease (ILD) | Dry cough/dyspnea/decreased lung function | Free payload is deposited on the alveolar epithelium | 5–15% (some ADCs) | Fatal → FDA clinical hold |
| Hepatotoxicity | Elevated transaminases/bilirubin | Kupffer cells phagocytose ADC → Payload release | 10–30% | Liver dysfunction → Prolonged hospitalization |
| Peripheral neuropathy | Numbness/pain in hands and feet | Toxicity of free payload to neuronal axonal microtubules | 10–20% (MMAE class) | Irreversible damage → Permanent discontinuation |
4.1.2 Reflections on the Q2 2026 Clinical Hold Case Involving Severe Off-Target Effects
The ADC clinical hold that garnered the most industry attention in Q2 2026 stemmed from a Claudin 18.2-targeted ADC.The drug demonstrated remarkable efficacy in a Phase II clinical trial for gastric cancer—with an ORR of 45% and a median PFS exceeding 7 months—but this success came at a cost: 31% of patients developed varying degrees of ILD, including 5% with Grade ≥3 severe ILD, and one patient died from ILD complications. The FDA immediately issued a partial clinical hold.
Post-hoc analysis sent a chill through the entire ADC community: the linker used in this ADC was not inherently flawed in design—it was a Val-Cit (valine-citrulline)-based enzyme-cleavable linker that, in theory, was only cleaved by cathepsin B within tumor cells.However, the crux of the problem lay in the bystander effect of the payload: after the ADC was cleaved in the tumor microenvironment and released the free payload, a portion of the payload was not fully absorbed by tumor cells but instead leaked from the tumor tissue into the bloodstream, where it was carried by the blood flow to the lungs and deposited there.This case taught the entire industry a costly lesson: ligand design can no longer be limited to the single criterion of “whether or not it cleaves in the bloodstream.”
4.2 Analysis of Cutting-Edge Technologies at CRS 2026
4.2.1 Evolution of Smart Responsive Linkers: The Design Logic Behind “Multi-Gate” Cleavage Mechanisms
If the evolution of ADC linkers from 2015 to 2020 was a single-track progression “from chemical cleavage to enzymatic cleavage,” then the period from 2024 to 2026 represents a multidimensional upgrade “from single-gated to multi-gated” systems.Traditional Val-Cit linkers rely on a single enzyme (Cathepsin B) as the cleavage trigger—this enzyme is highly expressed in various tumor cells, enabling efficient cleavage. However, the issue is that Cathepsin B is also expressed in certain normal tissues (such as liver Kupffer cells and osteoclasts), and free Cathepsin B released into the bloodstream from necrotic tumor tissue can non-specifically cleave circulating ADCs.This is the inherent flaw of single-gated systems.
The design philosophy behind multi-gated linkers is “don’t let the cat out of the bag until the rabbit is in sight”: the payload is released only when multiple conditions are met simultaneously. The most typical example is the “pH + enzyme” dual-gated linker.This type of linker contains a chemically sensitive group (such as an acetal or protonate) that is sensitive to slightly acidic pH (4.5–5.5) and an enzyme-cleavable sequence (such as Val-Cit or Val-Ala). In the bloodstream (pH 7.4), the pH-sensitive group remains stable and will not cleave even upon contact with free proteases—the first gate remains locked.After the ADC is internalized by the cell, the endosome acidifies to pH 5.0–6.0, causing the pH-sensitive group to become unstable—the first gate unlocks. Subsequently, cathepsin B in the lysosome cleaves the cleavage sequence—the second gate opens as well. The payload is finally released.
By 2026, multi-gate designs had evolved into triple-gate systems: pH + enzyme + reduction.Why introduce a reduction condition? Because the concentration of reduced glutathione (GSH) inside tumor cells is 100–1,000 times higher than in the blood. By embedding a disulfide bond—sensitive to reducing environments—into the linker, the conditions for payload release become: pH 4.5–5.5 + presence of cathepsin B + high GSH concentration—all three conditions are indispensable.This design has been proposed in multiple academic papers and patents from 2025–2026, and at CRS 2026, scientists from Seagen/Pfizer and Daiichi Sankyo are expected to present the first in vivo stability and antitumor activity data for triple-gated linkers in non-human primates (NHPs).
| Gate Types | Triggering Conditions | Specificity Source | Representative Conjugates | Technology Maturity |
| Single-gate (enzyme) | Cathepsin B cleaves Val-Cit | Cathepsin B is highly expressed in tumors | VC-PABC (Seagen/Pfizer) | Already on the market (Adcetris/Padcev, etc.) |
| Single-gate (chemical) | Hydrolysis of the imine bond at acidic pH | Slightly acidic tumor microenvironment | BR96-DOX (early-stage ADC) | Discontinued (unstable) |
| Dual-gate (pH + enzyme) | Pre-activated at acidic pH → Enzymatic cleavage | Endosome + lysosome dual barrier | Multiple biotech pipelines | Phase I/II clinical trials |
| Triple-gate control (pH + enzyme + GSH) | Acidic pH + Cathepsin B + High GSH | Triple verification → Extremely low off-target cleavage rate | First-in-human data at CRS 2026 | Preclinical/NHP Validation |
4.2.2 Quantitative Measurement of Macromolecular Stability in Vivo: New Analytical Tools for Real-Time Monitoring of Conjugated Drug Integrity
Advances in ADC linker design are inseparable from advances in analytical technology. At the CRS conference in 2026, a key sub-topic that cannot be overlooked is quantitative analytical methods for ADC in vivo integrity. Why is this topic so important? Because to this day, we still lack a standardized method capable of cheaply and efficiently distinguishing between intact ADC molecules, antibody + partial payload, and completely naked antibodies in clinical blood samples.
Currently, the most widely used method for in vivo ADC analysis is affinity capture LC-MS/MS: using anti-payload antibodies or anti-human IgG antibodies to capture all molecules containing payload or human antibodies from patient plasma, followed by mass spectrometry to analyze the presence and quantity of the payload.However, this method has two major drawbacks: first, it can only detect total payload and cannot distinguish whether the payload is attached to a complete ADC or has already detached; second, its sensitivity is insufficient—when the concentration of free payload in plasma is at the pg/mL level (which is precisely the concentration that causes pulmonary toxicity), conventional LC-MS/MS may fail to detect it at all.
A technical highlight at CRS 2026 was the application of fluorescence resonance energy transfer (FRET) in vivo imaging for ADC integrity tracking. The principle is simple: a FRET donor fluorophore is labeled on the ADC antibody, and a FRET acceptor fluorophore is labeled on the payload.When the payload is still bound to the ADC, the donor and acceptor are in close proximity, resulting in a strong FRET signal; once the payload is cleaved and released, the donor and acceptor separate, and the FRET signal disappears. By monitoring the decay curve of the FRET signal in real time, the rate constant of payload dissociation from the ADC in vivo can be precisely calculated. In mouse models, this technique achieves spatiotemporal resolution at the minute level.
4.3 The Perspective of the Business Decision-Making Level (Executive Thinking)
4.3.1 Expanding the Therapeutic Window Offers Greater Commercial Value Than Switching to a New Target
Consider this thought experiment: Suppose you are the head of business development at a multinational pharmaceutical company, and in Q2 2026, you have two companies to evaluate.Company A has an exciting new target—a membrane protein expressed only in a few cancer types—and preclinical data show excellent ADC efficacy. However, their linker uses technology that is twenty years old. Company B is working on HER2, an old target that has been thoroughly explored—but their linker is triple-gated, and in non-human primates (NHPs), the toxin release rate is only one-tenth that of traditional linkers. Which one would you choose?
Anyone who has worked in the pharmaceutical industry—or attended enough pharmaceutical conferences to see the pattern—will tell you: choose Company B. There are three reasons for this.First, while HER2 is an old target, it has been thoroughly validated by clinicians and regulators worldwide—you don’t need to spend five years and $200 million on target validation and proof-of-concept (PoC). Second, the market for HER2-targeted drugs (such as Trastuzumab and Enhertu) is already mature—doctors, patients, and payers all understand what HER2-positive means, so the cost of education is zero.Third, and most crucially—if Company B’s triple-gate linker can truly widen the safety margin (i.e., the gap between the effective dose and the toxic dose) by 3–5 times, that means an HER2 ADC like Enhertu—which has been shown to carry a serious risk of ILD—suddenly becomes a “super HER2 ADC” with significantly improved safety.
The business logic here is simple: in a mature, multi-billion-dollar market, upgrading a clinically accepted product into a safer version through delivery technology is far more profitable than building clinical awareness and market channels from scratch for an entirely new target. This is why, in M&A deals within the ADC sector from 2025 to 2026, linker platforms are more favored by capital than pipelines targeting new targets.As an anonymous Wall Street biotech analyst wrote in a Q2 research report: “The linker is the new target.”
4.3.2 Conference Guide: Skip the flashy efficacy slides and head straight to the poster session to examine free toxin kinetics
If you just sit in the main CRS conference hall listening to presentations, you’ll likely be moved by scenes like this: the presenter slides to a “urine cap” image—showing the tumor shrinking from baseline size to near invisibility—accompanied by the words “Complete Response,” followed by a round of applause from the entire audience.But as a knowledgeable attendee, you should ask yourself a question at this moment: Did the tumor shrink because of the ADC’s precision targeting, or because of the indiscriminate attack by free toxin—and did this tumor shrink at the cost of the patient simultaneously developing severe neutropenia and interstitial lung disease?
This is why we say: Skip the flashy efficacy PowerPoints and head straight to the poster session to examine the free toxin metabolism curves. The reason is simple: Oral presentations in the main hall typically last only 12–15 minutes, so presenters naturally devote their limited time to the most impressive data—ORR, CR, PFS.In contrast, the plasma concentration-time curve for free payload usually appears only in the supplementary data of posters, or even just in the investigators’ notebooks. Yet it is precisely this curve that serves as the most critical indicator for determining whether an ADC linker is truly safe.
How do you interpret this? Once you’ve located an ADC-related poster in the poster session, skip the first image showing tumor shrinkage and look for a chart titled “Plasma Concentration of Free Payload” or “ADC Pharmacokinetics.” If the free toxin curve appears as a flat, low-level line—indicating that the linker remains virtually intact in the bloodstream—then this ADC’s linker technology is worth paying attention to.If the curve you see is a parabola that rises sharply and then declines slowly—indicating that a large amount of toxin is released into the bloodstream within the first 24–72 hours after administration—the linker in this ADC is essentially a time bomb, posing a significant safety risk to patients.
A more rigorous approach is to calculate a ratio: the percentage of free toxin AUC relative to total payload AUC. This ratio directly reflects the in vivo stability of the linker. Among currently marketed mainstream ADCs, this ratio typically falls within the 2%–10% range—meaning that even the best-performing linkers on the market still have at least 2% of the toxin circulating in the bloodstream in a “dissolved” state.If you see an ADC on a poster with a free toxin AUC percentage below 1%, that’s a strong signal—the linker technology offers breakthrough stability. If it exceeds 10%, you should be highly vigilant—because even if the tumor shrinkage data looks impressive, the hidden risk of off-target toxicity could erupt in larger-scale clinical trials.
So, as you stroll through the poster session at CRS 2026, keep this simple rule of thumb in mind: look at the free toxin curve first, then at the tumor shrinkage graph.Don’t be blinded by the glamour of the tumor shrinkage charts—those are merely the results, while the free toxin curve is the cause. Especially when you stop in front of a small biotech’s poster: if it features a clean, crisp PK plot of free toxin metabolism and the free toxin AUC is <1%, grab a business card immediately—this could be the most valuable signal you find at the entire conference.Conversely, if a company’s poster only shows tumor shrinkage data but makes no mention of free toxin PK—then its ligand is likely its Achilles’ heel, and they just don’t want you to see it.
| Perspectives on the Competition | Green Light (Worth Watching) | Red Light Warning (High Alert) |
| Percentage of Free Toxin AUC | <1% (Extremely stable linker) | >10% (severe ligand leakage) |
| Time to Cmax of Free Toxin | 7–14 days post-administration (synchronized with ADC metabolism) | 1–3 days post-administration (early linker cleavage) |
| Free toxin curve shape | Flat, low-level plateau | Peaked parabolic curve |
| Data transparency | Proactively displays PK charts with complete data | Displays only efficacy; PK data is missing or unclear |
| Incidence of ILD | <3% (a direct reflection of ligand stability) | >10% (a direct consequence of free toxin deposition) |
5.0 Cross-Industry Brainstorming at biotech events (Cross-pollination): When ADCs Meet Nucleic Acids and Peptides—The Convergence of DDS Technologies
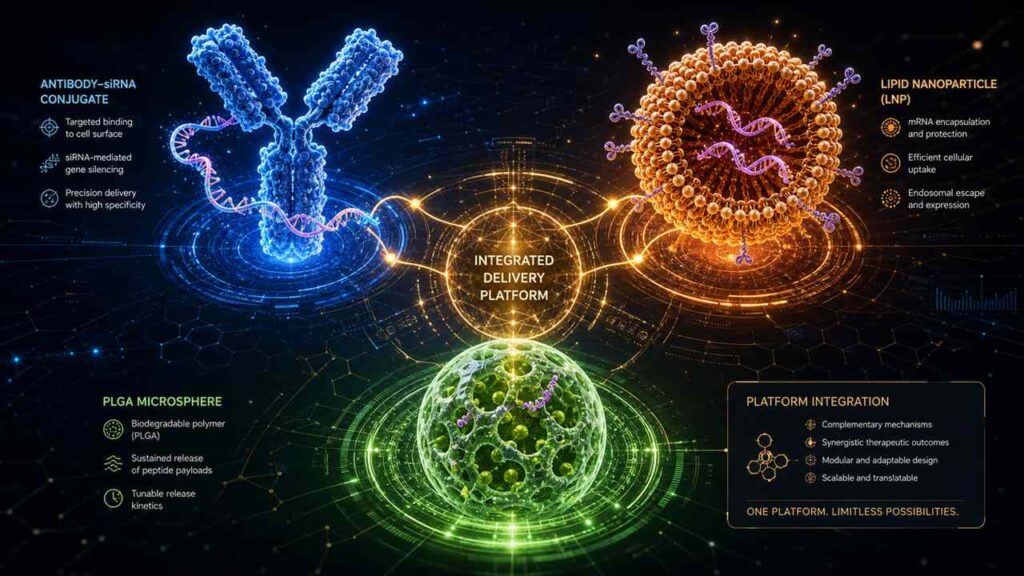
5.1 Potential Integration of AOCs (Antibody-Nucleic Acid Conjugates) and Microsphere Technology
5.1.1 Cross-Border Formulations: A Powerful Combination of Macromolecular Guidance and Nanodelivery
If we examine the three core DDS technology directions covered in the first four chapters—LNP-targeted delivery of in vivo CAR-T cells, long-acting sustained-release microspheres for GLP-1, and smart responsive linkers for ADCs—together, an exciting picture begins to emerge: these are no longer isolated vertical fields but are undergoing large-scale cross-fusion.
The first point of convergence is the AOC (Antibody-Oligonucleotide Conjugate). Traditional ADCs attach chemotherapeutic toxins to antibodies, whereas AOCs attach small nucleic acids (siRNA/ASO, i.e., antisense oligonucleotides) to antibodies. Why attach nucleic acids to antibodies?Because the biggest challenge currently facing small nucleic acid drugs is extrahepatic targeting—the vast majority of siRNA drugs are limited to hepatic delivery. However, if you conjugate an antibody capable of precisely targeting receptors on the surface of muscle cells (such as an anti-transferrin receptor TfR1 antibody) with an siRNA that can silence disease-causing mRNA in muscle tissue, you obtain a muscle-specific gene silencing drug.How significant is the commercial potential of this concept? It is vast enough to address a range of currently nearly untreatable neuromuscular diseases, including Duchenne muscular dystrophy (DMD), spinal muscular atrophy (SMA), and even amyotrophic lateral sclerosis (ALS).
The linker connecting the nucleic acid to the antibody in AOCs is essentially an ADC linker problem—you must ensure that this linker remains absolutely stable in the bloodstream and only cleaves to release the siRNA upon reaching the target tissue and being internalized by cells. The application of triple-gated linkers in AOCs offers even greater advantages than in ADCs, as nucleic acid molecules (siRNA) are more sensitive to chemical environments, requiring higher precision in linker cleavage.
The second point of convergence is the “microsphere + small nucleic acid” combination. If siRNA drugs can be encapsulated in degradable PLGA microspheres to achieve sustained release of small nucleic acids over a period of 3–6 months following subcutaneous injection, this would effectively upgrade small nucleic acid drugs from a quarterly injection regimen to a long-acting formulation requiring only an injection every six months.For chronic conditions such as hypertension and hyperlipidemia, a semi-annual RNAi injection would represent an ideal treatment regimen that causes virtually no disruption to patients’ daily lives. However, the technical challenges are evident: siRNA molecules are more fragile than peptides, and the acidic environment inside PLGA microspheres is fatal to RNA degradation. This necessitates the incorporation of a dual-buffer system within the microsphere matrix.
The third intersection—and the one with the most futuristic potential—is LNP-delivered AOC. This approach does not attach nucleic acids directly to antibodies; instead, nucleic acids are encapsulated within LNPs, which are then conjugated to antibodies on their surface. The benefits of this method are twofold: LNPs provide extremely high nucleic acid loading capacity (a single LNP can carry thousands of mRNA or siRNA molecules), while antibodies offer precise cellular targeting capabilities.The combination of these two modules could, in theory, increase the number of siRNA copies received by target cells by 1–3 orders of magnitude.
| Cross-fusion technology | Combination Methods | Core Advantages | Key Technical Bottlenecks | Representative Pipelines |
| AOC | Antibody + siRNA/ASO (Antibody-Targeted) | Extrahepatic Targeting (Muscle/CNS/Tumor) | Ligand in vivo stability + nucleic acid loading capacity | Avidity/Dyne Tx (clinical stage) |
| Microspheres + small nucleic acids | PLGA microspheres encapsulating siRNA | 3–6 months of sustained release | Acid-induced RNA degradation + RNase | Multiple academic laboratories (preclinical) |
| LNP-AOC | Antibody-modified LNP encapsulating nucleic acids | Dual advantages of high loading capacity and precise targeting | Stable conjugation of antibodies to the LNP surface | Early-stage academic research |
| Microspheres + In vivo CAR-T | PLGA Microspheres for Sustained-Release CAR mRNA LNP | Sustained Generation of CAR-T Cells | Precise control of time-dependent release | Conceptual Phase |
5.2 The Logic Behind the Success of Platform Technologies
5.2.1 Why Can Breakthroughs in Underlying Delivery Platforms Trigger a “Catch-All Effect”?
If you look back at the history of mergers and acquisitions in the biopharmaceutical industry over the past two decades, you will notice a recurring pattern: the most valuable companies are never those that have developed a single successful drug, but rather those that own a robust platform. Moderna was valued at billions of dollars even before the COVID-19 pandemic—not because of any specific drug in its pipeline, but because it possessed an LNP platform capable of rapidly designing, synthesizing, and testing any mRNA drug.Alnylam’s small-nucleic-acid empire—from Onpattro to Givlaari to Oxlumo—is built entirely on the same GalNAc-siRNA liver-targeting delivery platform.
The value of “platform thinking” is particularly evident in the field of drug delivery systems (DDS), as DDS is inherently a reusable infrastructure.Once you’ve solved the fundamental problem of “how to efficiently deliver mRNA to T cells,” your platform can immediately be repurposed to deliver any mRNA to any lymphocyte of similar size—CAR-T therapy for lymphoma, CAR-T therapy for autoimmune diseases, mRNA vaccines to activate anti-tumor T-cell immunity, gene editing to correct immune defects in T cells… One platform, ten pipelines, and a commercial potential worth tens of billions of dollars.
This is why BI is willing to spend $2 billion to acquire an in vivo CAR-T LNP platform—it’s not buying a single drug, but a foundational access pass that can be continuously repurposed.Similarly, this is why at CRS 2026, you should pay close attention to biotech companies that aren’t just presenting clinical data for a single pipeline, but are systematically demonstrating that “our platform has validated delivery efficiency across multiple cell types (T cells, B cells, hematopoietic stem cells) and multiple animal models (mice, rats, NHPs).”
6.0 On the Ground in Lisbon: A Practical Guide to Attending biotech events (and Online Tracking) CRS 2026

6.1 “Most Efficient Networking and Information Filtering Strategies” for an Event with 4,200 Attendees
6.1.1 Using the Official App to Filter Closed-Door Sessions Optimized for ADC Linkers and Extrahepatic LNPs
At a conference with 4,200 attendees, if you go in with the mindset of just wandering around and looking at things, you’ll likely spend all your time sifting through irrelevant posters and booths, and end up remembering nothing. As someone with clear conference objectives, you need a “sniper-style” strategy—precise targeting, rapid acquisition, and decisive exit.
For a Lisbon biopharma summit of this scale, the official CRS 2026 app is your most important tool. While the vast majority of attendees use it only to view the schedule and map, you can enable the keyword filter immediately after registration and set the following keywords: LNP targeting, extrahepatic, siRNA delivery, biodegradable linker, multi-gated, PLGA microsphere, burst release, transcellular permeation enhancer, oral peptide, free payload PK, ADC stability.The app will automatically filter out oral presentations, posters, and satellite sessions containing these keywords. Spending 15 minutes each morning planning your route for the day based on this filtered list is 10 times more efficient than blindly wandering between venues with the crowd.
For key topics of interest—in vivo CAR-T LNPs and ADC linkers—we recommend prioritizing Concurrent Session Oral Presentations and Rapid Fire Presentations. These are typically where the latest, cutting-edge data is first disclosed.While Plenary Lectures feature high-profile speakers, the content is often review-based material that has been presented multiple times elsewhere. As for the Exposition: the booths of large pharmaceutical companies are incredibly lavish, but the information density is surprisingly low—as anyone with experience knows, the staff at these booths are mostly from marketing and PR teams, and their technical depth rarely goes beyond the level of a product brochure.You should allocate 80% of your time in the exhibition hall to the modest booths tucked away in the corners of small biotech companies—the kind with just a table, a laptop, and two nervous postdoctoral researchers.
| Types of Information Sources | Information Depth | Freshness of Information | Commercial Bias | Target Audience | Recommended Time Allocation |
| Plenary Session | Low to moderate (primarily reviews) | Medium (mostly published works) | Low | Beginners/General Audience | 10% |
| Oral presentation in a breakout session | High (latest data) | Very high (first disclosure) | Medium | Professional BD/Investors | 30% |
| Rapid Fire | Medium-High (Key Metrics) | Very High (Initial Release) | Low to medium | BD/Investors | 20% |
| Poster Session | Highest (Raw Data) | Very high | Low | In-depth Technical Evaluators | 30% |
| Small Biotech Booth | High (one-on-one interactions) | High | Medium (sales pitches present) | BD prospecting | 10% |
6.2 The Art of Asking Questions: How to Get to the Heart of the Matter During Q&A Like an Industry Veteran?
6.2.1 Avoid Vagueness: Challenge Speakers with Plain Language and Hard Data
The Q&A session is the true test of a conference. Anyone can deliver a polished presentation, but when suddenly asked a pointed technical question during Q&A, the speaker’s response is the best indicator of a team’s true capabilities. Below are several killer question templates I’ve prepared for you, covering the three core topics discussed in this article—feel free to use them directly in the relevant breakout sessions.
For presenters on in vivo CAR-T LNP delivery: Your biodistribution data shows X% of the LNP dose accumulating in the spleen. Can you break that down further—what fraction of the spleen-localized LNP is actually taken up by T cells versus other splenic cell types like macrophages and dendritic cells? And have you quantified the absolute number of CAR mRNA molecules per T cell at your therapeutic dose?
For the ADC linker reporter: You showed impressive tumor regression data, but I noticed the free payload plasma concentration was not reported. Can you tell us the Cmax and AUC of the free payload? Specifically, what is the safety margin—the ratio of the toxic dose free payload AUC to the therapeutic dose free payload AUC?What is the safety margin?)
To the presenter of the long-acting GLP-1 microspheres: Your in vitro zero-order release profile looks excellent over 30 days. But when you scale up from lab to pilot batch, how does the burst release percentage change? And what is your strategy to control the acidic microclimate inside the degrading PLGA microspheres?
Each of these questions directly addresses the most critical pain points—the data that’s often buried in the fine print on the last page of supplementary materials—for this technology. If you can ask these questions during the Q&A session, the presenter on stage will immediately recognize you as an expert in the field, and the audience around you will give you a knowing look—well asked.
7.0 Conclusion from biotech events: DDS Is No Longer a Supporting Role; It Is the Crown Jewel of the Biopharmaceutical Value Chain Post-2026
7.1 Summary: The Internal Logical Loop from Q2 Hot Topics to the CRS Annual Meeting
7.1.1 Drug Delivery Systems (DDS) Are the Skeleton Supporting the Battle for Cutting-Edge Biopharmaceuticals
Let’s return to those thrilling 48 hours in early June 2026: Legend Biotech used a small-sample Phase I dataset to demonstrate that CAR-T cells can be generated in situ within the human body; BI bet $2 billion that SLE would become the first “consumer-grade” indication for in vivo CAR-T; Alnylam announced the absolute dominance of small nucleic acids in the chronic disease sector with 18-month long-acting lipid-lowering data;Arrowhead used CNS delivery data in non-human primates to break new ground in extrahepatic small-nucleic-acid targeting.
These four events may appear unrelated, but they share a single underlying driving force: breakthroughs in drug delivery systems (DDS). Without T-cell-targeting LNPs, in vivo CAR-T would remain nothing more than a beautiful academic concept. Without GalNAc conjugation and novel ligand chemistry, small nucleic acids would forever be trapped in the liver. Without smart responsive linkers, ADCs could not overcome the safety ceiling of off-target toxicity.Without the controlled release of PLGA microspheres and the advancement of oral permeability enhancers, GLP-1 would remain nothing more than a weekly injection for the wealthy seeking weight loss, rather than a universal chronic disease management solution that can be taken orally.
Walk the halls of any drug delivery conference today, and the message is unmistakable: DDS has evolved from a supporting role in drug development to the crown jewel of the biopharmaceutical value chain. In the past, delivery was considered synonymous with formulation—the final step in a product’s development, a packaging task casually handled by the formulation department during the CMC phase. But in 2026, this perception is completely outdated.DDS is now a direct creator of clinical value—it not only influences a drug’s pharmacokinetics but also directly determines the upper limits of its efficacy and safety, and directly defines whether a therapeutic approach has the potential for commercialization.
7.2 The Ultimate Forecast
7.2.1 Whoever masters the next generation of safe, precise drug delivery systems will hold the pricing power in the biopharmaceutical industry for the next decade
At this juncture in 2026, the global biopharmaceutical industry is undergoing a profound restructuring of its value chain. Twenty years ago, a pharmaceutical company’s core competitiveness lay in discovering new targets—the target was everything.Ten years ago, core competitiveness expanded to include the R&D pipeline—companies with several Phase III pipeline candidates in hand were the winners. By 2026, the industry’s competitive logic has evolved once again: targets and pipelines are merely raw materials, while the true core competitiveness has shifted to delivery infrastructure.
The essence of this shift in logic is this: as cutting-edge biopharmaceuticals transition comprehensively from small chemical molecules to macromolecules (antibodies/ADCs), nucleic acids (mRNA/siRNA/ASO), and live-cell therapies (CAR-T/stem cells), these novel molecules share a common characteristic—they are either too large or too fragile to cross the cell membrane and reach their targets on their own, as aspirin does.You need a delivery vehicle. And the quality of the delivery vehicle you possess—its targeting precision, delivery efficiency, safety margin, and controlled-release capabilities—determines the width of your commercial moat.
Therefore, my ultimate prediction for the next decade is this: whoever masters the next generation of safe, precise drug delivery systems will hold the pricing power in biopharmaceuticals for the next ten years. Not because the delivery system itself can be sold for a high price, but because it serves as an “enabling layer”—any new drug seeking to enter the field of in vivo cell or gene therapy must travel along the delivery highway you’ve built and pay you a toll.This is why MNCs are willing to spend billions of dollars to acquire a biotech platform that focuses solely on delivery—they are not buying a product, but rather a ticket to access the infrastructure for all potential next-generation biologics over the next decade.
July 2026, Lisbon. Among the life science events set to define the pharmaceutical calendar, the CRS Annual Meeting will serve as the first global showcase of this delivery infrastructure war. We’ll be watching closely.
8.0 biotech events FAQ: Five Frequently Asked Questions About In Vivo CAR-T, DDS, and the CRS 2026 Annual Meeting

Question 1: How much of a difference is there really in efficacy between in vivo CAR-T and traditional in vitro CAR-T?
This is one of the most frequently asked questions in the industry, yet also one of the hardest to answer directly, as clinical data on in vivo CAR-T remains very limited (currently only Legend Biotech’s Phase I data). Based on existing real-world data for ex vivo CAR-T, the best complete remission (CR) rate for DLBCL is approximately 50%–60%, but this is from a strictly selected, ideal patient population.Phase I data for in vivo CAR-T show a CR rate of 33%. Given that this was observed in the lowest dose group—where Phase I doses are typically far below the optimal dose—this figure is encouraging. More importantly, the safety profile of in vivo CAR-T (no Grade ≥3 CRS or ICANS) allows for greater flexibility in dose escalation—a limitation that often prevents further dose increases in ex vivo CAR-T due to CRS/ICANS concerns.Therefore, a reasonable expectation is that, at the optimal dose in Phase II trials, in vivo CAR-T is expected to achieve a CR rate on par with or even surpassing that of ex vivo CAR-T, while offering an order-of-magnitude advantage in terms of safety. However, all of this hinges on the continued improvement of LNP delivery efficiency.
Question 2: When will small nucleic acid therapies targeting extrahepatic sites enter clinical use?
Based on currently available pipeline information, the most optimistic estimate is 2028–2029. Arrowhead and Alnylam’s CNS-targeted pipelines are currently in the preclinical/NHP validation phase, with IND submissions expected in 2026–2027; Phase I clinical trials typically take 18–24 months.If all goes smoothly, the first small interfering RNA (siRNA) drug targeting sites outside the liver (most likely a CNS-targeting pipeline for Huntington’s disease or ALS) may yield initial in-human efficacy data around 2029 and enter pivotal clinical trials between 2030 and 2032. However, it is important to note that the safety threshold for CNS delivery is extremely high—any unexpected neurotoxicity would result in the immediate termination of the pipeline.Consequently, industry insiders tend to believe that muscle-targeted small nucleic acids (such as AOC for treating DMD) may reach the market sooner than CNS-targeted therapies, as safety risks in muscle tissue are relatively manageable.
Question 3: Which will be the ultimate winner—the “once-monthly” GLP-1 microsphere injection or the oral tablet?
The answer may not be an either-or choice, but rather a tiered coexistence. From a market analysis perspective, once-monthly long-acting injections are likely to become the clinical treatment of choice for type 2 diabetes and severe obesity (BMI ≥ 35)—because these patients require continuous GLP-1 receptor activation, and a monthly regimen imposes a sufficiently low burden on patient compliance.Meanwhile, oral tablets (especially next-generation products that are not affected by meals) will dominate the larger consumer market for mild-to-moderate weight loss and metabolic health—targeting the sub-healthy population with BMIs between 27 and 32 who have psychological barriers to injections and less urgent weight loss needs.These two markets are not only non-conflicting but complementary—oral tablets are responsible for user acquisition (attracting users who previously hesitated to use injections), while long-acting injections are responsible for retention (providing an upgrade option for users requiring greater efficacy).
Question 4: Which three biotech companies deserve the most attention at the 2026 CRS Annual Meeting?
Based on pre-conference abstract leaks and Q2 industry trends, the following three biotech companies deserve close attention: (1) Company N, which specializes in T-cell-targeted LNP delivery—it is expected to announce data on the spleen T-cell transfection efficiency of its SORT-LNP in NHP models, representing a breakthrough in overcoming a key bottleneck for the transition of CAR-T from academia to industry.(2) Company L, specializing in triple-gate ADC linkers—its pH+enzyme+GSH triple-responsive linker has demonstrated remarkable plasma stability in preclinical studies (free payload AUC < 0.1%); if NHP data are replicated, it could become the benchmark for next-generation ADC linkers.(3) Company R, specializing in oral microneedle capsules—a spin-off from MIT’s Langer/Traverso team—has advanced its insulin microneedle capsule into Phase I clinical trials, and preclinical data for the GLP-1 version may be disclosed for the first time at CRS.
Question 5: As a non-specialist (investor/BD), what is the most important thing to prepare in advance for the CRS Annual Meeting?
Prepare a three-question checklist. The CRS conference is extremely information-dense; the only way to stay focused in a noisy environment with 4,200 attendees is to go in with very specific questions. We recommend spending a day before departure preparing three specific technical questions for each of three core topics (refer to the Q&A template in Chapter 6 of this article), printing them on a single sheet of paper, and carrying it with you.After every oral presentation or poster session, ask yourself the questions and write down your answers in the corresponding spaces on your three-question checklist. If a company or lab’s presentation allows you to mark all three questions with a satisfactory answer—then that’s the one you should focus on, no matter how small their booth is or how low their stock price is.





