

- 1. The ESC Congress 2026, a pivotal bio meeting 2026, is more than just a cardiovascular conference; the real question following GLP-1 is whether “long-term cardiovascular value can be demonstrated.”
- 2. At the bio meeting 2026, the expansion of AOCs and small nucleic acids beyond the liver has raised a new question for the cardiovascular field: Can delivery platforms truly be reused across different tissues?
- 3. At the bio meeting 2026, GLP-1, AOC, and new cardiovascular drugs all point to one change: clinical narratives must withstand scrutiny from manufacturing and quality systems
- 4. bio meeting 2026: ESC Congress Attendance Strategy for Different Priorities for BD, R&D, and Investment Teams
- 5. The Most Valuable Takeaway from the bio meeting 2026 Is Not Conclusions, but a Set of Judgment Questions
- 6. Pre-Conference Checklist: Turning the bio meeting 2026 into an Effective Information Screening Experience
- 7. bio meeting 2026 FAQ: Search-based Questions to Address When Writing About the ESC Congress

1. The ESC Congress 2026, a pivotal bio meeting 2026, is more than just a cardiovascular conference; the real question following GLP-1 is whether “long-term cardiovascular value can be demonstrated.”
The significance of the ESC Congress 2026, a pivotal bio meeting 2026, extends far beyond that of a typical annual cardiovascular conference. It is becoming the central venue for validating the commercial value of new drugs in the fields of metabolism, obesity, heart failure, and cardiovascular risk management. Here, pharmaceutical companies can see a complete chain of evidence, from clinical endpoints to market pricing. The European Society of Cardiology Annual Congress will take place in Munich, Germany, from August 28 to 31, 2026.Official website: https://www.escardio.org/Congresses-Events/ESC-Congress. According to 2025 data, the conference attracted over 33,300 attendees, including more than 29,300 healthcare professionals on-site and over 4,000 online participants from 169 countries worldwide.Given this scale, any major study presented during the “Hot Lines” session will find its way onto the discussion tables of global cardiovascular clinical practice guidelines within 48 hours.
However, the conference’s sheer size is not what attendees should truly be focusing on. The core tension of the 2026 conference lies in the fact that GLP-1 agonists have already gained market entry through weight-loss data; the next question is whether they can produce sufficiently robust evidence of cardiovascular outcomes to transform themselves from a “metabolic blockbuster” into a “cardiovascular essential.”If this transformation materializes, the market ceiling for semaglutide and tirzopotide will be redefined; if it fails to hold up, the valuation premium across the entire GLP-1 sector will face downward pressure.
Meanwhile, the intersection of these two storylines—highlighted by Novartis’s $12 billion acquisition—is precisely the question the pharmaceutical industry most urgently needs to answer at this biopharma summit in 2026. One is the validation of GLP-1’s cardiovascular value; the other is whether nucleic acid drug platforms can break into cardiovascular indications. AOCs (antibody-oligonucleotide conjugates) and small nucleic acid drugs have entered the cardiovascular spotlight.The shift of nucleic acid therapeutics from liver-targeted applications to the myocardium, skeletal muscle, and even the central nervous system signifies a transformation in the development landscape of cardiovascular drugs. However, the fervor surrounding platform deals does not equate to the ability to bring products to market; validation of delivery efficiency, safety, tissue selectivity, and long-term administration is essential—not a single aspect can be overlooked. The ESC Congress 2026 thus carries dual significance: one is the Avidity event, which brought together
over 33,300 attendees, including more than 29,300 healthcare professionals on-site and over 4,000 online participants from 169 countries worldwide. The 2026 event is expected to maintain or slightly exceed this scale.The official website for the conference is https://www.escardio.org/Congresses-Events/ESC-Congress. Although it is a clinical medical society, the premiere of findings at the Hot Lines sessions has established it as the premier global stage for evaluating the commercial value and clinical efficacy of new GLP-1 drugs. For pharmaceutical companies, CDMOs, and investors in the cardiovascular and metabolic fields, missing the ESC Congress means missing the most critical window of opportunity to gauge industry trends for the year.All recent clinical trials of GLP-1 drugs in the areas of cardiovascular weight loss, heart failure, and metabolic syndrome (such as the SELECT study) have been presented at this conference. The ESC Congress 2026 will be held in Munich, Germany, from August 28 to 31, 2026. The exact attendance figure for 2025 was
It would be a misjudgment to view the ESC Congress 2026 as just another cardiovascular conference Munich. Its value lies not only in attendance numbers and clinical influence but also in its continued role as the central venue for validating the commercial value of new drugs in the fields of metabolism, obesity, heart failure, and cardiovascular risk management.The research data released annually on this platform directly influences the direction of updates to ESC guidelines, the evidence base for health insurance negotiations in various countries, and the prioritization of pharmaceutical companies’ pipelines. For companies operating in the cardiovascular and metabolic field, the ESC Congress serves as both a window for information gathering and a pivotal point for strategic recalibration.
1.1 From “Weight-Loss Drugs” to “Cardiovascular Outcome Drugs”: GLP-1 Is Being Repositioned
The clinical narrative surrounding GLP-1 receptor agonists has undergone a fundamental shift. Over the past three years, market enthusiasm for semaglutide and tirzopotide was primarily driven by weight loss efficacy—the weight loss curves demonstrated in the STEP series of clinical trials were sufficient to make Wegovy and Zepbound phenomenon-level products.However, what truly influences pharmaceutical companies’ business development decisions and investors’ judgments is never the weight loss figures themselves; pricing is never determined by those numbers alone. The four key factors that truly shape these decisions are: whether hard endpoints are met, whether indications can be expanded, whether patient stratification is clear, and whether the reimbursement logic holds up.
The SELECT trial marked a pivotal turning point. This was the first GLP-1 drug to demonstrate a reduction in the risk of major adverse cardiovascular events (MACE), and it was approved based on cardiovascular outcomes rather than glycemic control or weight loss. From a pricing perspective, the approval of the cardiovascular indication means that GLP-1 is no longer merely a “weight-loss option” but has entered the reimbursement framework for cardiovascular prevention, which has a structural impact on its commercial ceiling.Semaglutide reduces the risk of major adverse cardiovascular events (MACE) by 20%. This data directly led to the U.S. FDA’s approval of Wegovy’s cardiovascular indication in March 2024. The study, involving 17,604 overweight or obese adults with diagnosed cardiovascular disease but no diabetes, demonstrated that a once-weekly subcutaneous injection
| Trial Name | Drug | Number of Participants | Primary Endpoint | Results | Impact on Commercial Pricing |
| SELECT | Semaglutide | 17,604 | MACE (cardiovascular death + non-fatal myocardial infarction + non-fatal stroke) | 20% reduction in MACE risk | Approved by the FDA in March 2024 for cardiovascular indications, qualifying for the CV prevention reimbursement framework |
| STEP-HFpEF | Semaglutide | 529 | KCCQ-CSS Score + Weight Change | Symptom improvement + weight loss | Expanded to include patients with HFpEF (heart failure with preserved ejection fraction) |
| SURPASS-CVOT | Telopectin | 13,383 | MACE (cardiovascular death + non-fatal myocardial infarction + non-fatal stroke + coronary revascularization) | Results to be released in 2025; non-inferior to semaglutide | Impact on the competitive positioning of dual-target GLP-1/GIP drugs in the cardiovascular market |
| FLOW | Semaglutide | 3,577 | Composite renal endpoint (decrease in eGFR + renal replacement therapy + renal death) | Study terminated early due to significant efficacy | Opening up opportunities for chronic kidney disease indications and expanding systemic value |
Each trial listed in the table above represents a drug’s journey from “proven efficacy” to “reimbursement,” a process involving a comprehensive set of patient stratification, guideline recommendations, and health insurance negotiations.SELECT demonstrated cardiovascular outcomes, marking an expansion of the STEP-GLP-1 commercial landscape. Semaglutide. However, the data itself is merely the starting point; what truly determines pricing is whether these data can persuade payers to accept a broader prescribing scope. Can telapreotide catch up in the cardiovascular market? HFpEF targets heart failure, FLOW targets kidney disease, and SURPASS-CVOT will determine
The questions attendees should be asking are very specific: Do the cardiovascular benefits extend across different BMI subgroups? Is performance consistent in patients with comorbid diabetes? Are the data from the heart failure subgroup sufficient to support an application for an independent indication? Are there any updates on long-term safety data (particularly regarding pancreatitis and gastrointestinal adverse events)? How should weight regain and the resurgence of cardiovascular risk following discontinuation be interpreted?How well these questions are answered will directly determine the pricing potential for GLP-1 agonists over the next three years.
The SELECT study transformed the pricing foundation for GLP-1 drugs. This study enrolled 17,604 overweight or obese patients with a history of cardiovascular disease and demonstrated that semaglutide reduced the risk of MACE by 20%.This finding transformed GLP-1 agents from adjunctive antidiabetic therapies into cardiovascular protective agents, and the pricing logic shifted accordingly—payors began to reassess their cost-effectiveness. If the evidence for cardiovascular hard endpoints is sufficiently strong, GLP-1 agents may be included in broader reimbursement coverage for cardiovascular disease prevention.
However, the boundaries of efficacy generalization remain unclear. 83%. For younger patients with isolated obesity, those with heart failure with preserved ejection fraction, or those with end-stage chronic kidney disease—the SELECT study excluded patients who had recently experienced a cardiovascular event, and the average age at enrollment was […]. Can the cardiovascular benefits of GLP-1 be proportionally generalized to these subgroups? These questions directly impact prescribing ceilings.Precisely these subgroup analyses will be the most noteworthy developments to track at the ESC Congress 2026. The average age was 61.7 years, and patients with diabetes accounted for approximately
From a payer’s perspective, the cost-effectiveness assessment of GLP-1 requires recalibration. If the cardiovascular outcome data are sufficiently robust, payers may include GLP-1 in the reimbursement scope for primary cardiovascular prevention. Conversely, if unexpected signals regarding long-term safety emerge—such as pancreatitis, thyroid C-cell hyperplasia, or functional decline due to loss of lean body mass—payers will tighten reimbursement criteria.The outcome of this tug-of-war will gradually emerge during the Hot Lines and Late-Breaking Science sessions at the ESC Congress 2026.
| Research | Primary Endpoint | Enrollment (N) | Reduction in MACE | Key Limitations |
| SELECT | 3P-MACE | 17,604 | 20% | Excludes recent CV events; high prevalence of diabetes |
| FLOW | Renal composite endpoint | 3,537 | To be released | Limited to diabetic nephropathy |
| STEP-HFpEF | KCCQ-CSS | 529 | N/A | Small sample size; non-hard endpoint |
Competitive strategies among pharmaceutical companies in the GLP-1 sector are diverging. Novo Nordisk is building its product portfolio through a multi-indication strategy for semaglutide, while Eli Lilly is doing the same via its own semaglutide portfolio, simultaneously advancing oral formulations and higher-dose pen injectors.Tirzopentide’s dual-target mechanism (GLP-1 plus GIP) aims to differentiate itself in terms of weight loss efficacy and directly competes with semaglutide in the SURPASS-CVOT cardiovascular outcomes trial. The core of the competition has shifted from which drug is more effective for weight loss to which can demonstrate broader systemic cardiovascular benefits.
The release of GLP-1-related substudies—such as analyses of heart failure phenotypes, renal function subgroups, or post-discontinuation rebound data—indicates that the academic community is moving beyond the framework of GLP-1 as a weight-loss drug and examining it within the context of the more complex management pathway for cardiovascular and metabolic diseases. Attendees should pay close attention to whether these substudies alter the position of GLP-1 in clinical practice guidelines.The agenda for the 2026 ESC Congress itself sends a signal. If multiple sessions in the Hot Lines section
In the transition from weight-loss drugs to cardiovascular outcome drugs, the reduction in MACE is significant, while the pricing of weight-loss drugs is based on the percentage of weight loss. When a drug possesses all three of these effects, payers must decide which dimension to use as the pricing benchmark. If cardiovascular protection is used as the basis for pricing, the price of GLP-1 could be significantly higher than that of comparable drugs priced based on glycemic control, as the economic burden of cardiovascular events is far greater than that of glycemic control.However, if payers insist on using weight loss efficacy as the basis for pricing, the value of GLP-1’s cardiovascular benefits will be underestimated. The pricing challenges facing GLP-1 are multidimensional. Traditional antidiabetic drugs are priced based on the reduction in HbA1c, while cardiovascular protection drugs are priced based on
Novartis announced in early 2026 that it had acquired the AOC platform, which already demonstrated the potential to progress from early-stage clinical trials to late-stage development. However, key factors to monitor regarding the AOC platform’s commercialization path include: how much of the acquisition price represents a platform premium; whether the CMC team will be able to maintain independent operations; and whether the conjugation process will be transferred to Novartis’s manufacturing system.Avidity Biosciences: This deal sends a clear signal to the industry—large pharmaceutical companies value the specific benefits of the DMD indication, and the pricing of options for expanded indications such as cardiovascular disease requires careful analysis by business development teams. For attendees of the ESC Congress, the progress of the post-merger integration—particularly Avidity’s
What truly influences the judgments of pharmaceutical companies and investors lies in hard endpoints, indication expansion, patient stratification, and reimbursement logic. Weight loss curves may appear impressive in clinical trials, but if they do not translate into a reduction in cardiovascular events, lower hospitalization rates, or sustained improvements in quality of life, their commercial value is limited.The SELECT study demonstrated a reduction in MACE rather than inferring cardiovascular benefits indirectly through weight loss. The pricing logic for GLP-1 drugs stems precisely from the fact that it directly demonstrated
The pace of indication expansion is also a key factor influencing the commercial value of GLP-1 in the SURPASS-CVOT context. From diabetes to obesity, from obesity to cardiovascular protection, and from cardiovascular protection to heart failure and CKD—each step in indication expansion requires specifically designed clinical trials and separate regulatory approvals.The pace of expansion depends on the rate of clinical data accumulation, the efficiency of regulatory communications, and pressure from competing products. If positive data are released at the ESC Congress 2026, it could accelerate the market positioning of GLP-1 dual-target drugs in cardiovascular indications and reshape the competitive landscape. Telporotide’s
| GLP-1 Indication Expansion Phase | Key Studies | Endpoint Type | Commercial Impact |
| Diabetes (Approved) | SUSTAIN Series | HbA1c | Target Market |
| Obesity (Approved) | STEP Series | Weight Loss (%) | Expanded Prescribing Population |
| Cardiovascular Protection (Approved) | SELECT | MACE (hard endpoint) | Rethinking Pricing Logic |
| Heart Failure (HFpEF) | STEP-HFpEF | KCCQ-CSS | New Indication Opportunities |
| Chronic Kidney Disease | FLOW | Renal Composite Endpoint | Nephrology Prescribing Pathways |
| MASH | HELIOS-SSL | Improvement in Liver Fibrosis | Entry into the Liver Disease Market |
1.1.1 The most critical question to ask in clinical practice is not whether the drug is effective, but whether its efficacy can be transferred to more complex patient populations
Whether efficacy can be transferred from “uncomplicated populations” to “complex populations” is a hurdle that GLP-1 must overcome to transition from a blockbuster drug to a routine prescription.The enrollment criteria for the SELECT trial were relatively straightforward—participants had a confirmed diagnosis of cardiovascular disease but no diabetes. While this design facilitates the detection of a clear efficacy signal, cardiovascular patients in the real world are far more complex than those in clinical trials. The combination of heart failure, diabetes, chronic kidney disease, advanced age, and polypharmacy—when these factors overlap—represents the true clinical scenario that prescribing decisions must address.
| Dimensions of Complex Patient Populations | Current State of Evidence | Translation Risks | Questions Attendees Should Ask |
| Concomitant HFpEF (Heart Failure with Preserved Ejection Fraction) | STEP-HFpEF demonstrated symptomatic improvement, but evidence on hard endpoints remains limited | Moderate: Symptom improvement does not equate to improved prognosis | Are there expanded outcome data for HFpEF? Are the results of subgroup analyses consistent? |
| Comorbid Chronic Kidney Disease | The FLOW trial was terminated early due to significant efficacy, with positive results | Low: Existing renal composite endpoints provide support | What is the safety profile across different eGFR strata? What is the dosing adjustment regimen for patients on dialysis? |
| Concomitant type 2 diabetes | Trials such as SUSTAIN-6 include data from diabetic subgroups, but were not originally designed for cardiovascular outcomes | Moderate: Background therapy is more complex in the diabetic population | What about the safety and efficacy of combination therapy with SGLT2 inhibitors? |
| Elderly patients (>75 years) | Limited subgroup data; concerns regarding the risk of muscle loss and malnutrition | High: Tolerability and adherence in the elderly population are uncertain | Are there specific subgroup analyses for the elderly? What is the discontinuation rate? |
| Long-term adherence | Real-world data show a 6-month discontinuation rate exceeding 30% | High: Adherence directly impacts the realization of cardiovascular benefits | Are there any comparative data on adherence between long-acting formulations and oral formulations? |
| Rebound after discontinuation | STEP 1 extension phase data show significant weight regain after discontinuation | Gao: Weight regain may offset cardiovascular benefits | Are there studies on maintenance therapy strategies? What about monitoring for cardiovascular events after discontinuation? |
Each row in this table corresponds to a real-world decision point that prescribing physicians encounter in clinical practice. Take adherence as an example: real-world data show that the 6-month discontinuation rate for GLP-1 agonists exceeds 30%. This means that even if the efficacy is proven, the cardiovascular benefits for a significant proportion of patients may be compromised due to treatment interruption.Another example is weight regain after discontinuation—data from the STEP 1 extension phase clearly show that body weight rebounds significantly after discontinuing semaglutide, yet there is currently a lack of sufficient long-term follow-up data to determine whether this rebound is accompanied by a renewed increase in cardiovascular risk.
The treatment paradigm for cardiovascular and metabolic diseases has shifted, with the core of this shift being a gradual transition in treatment goals from “glucose control” and “weight loss” to “reducing cardiovascular hard endpoints.”GLP-1 agonists are at the center of this shift. If the Hot Lines at the ESC Congress 2026 release more subgroup data from complex patient populations, it will directly accelerate or delay the pace of this transition. Attendees need to assess whether these data are sufficient to alter current prescribing practices, rather than simply taking note of positive results.
The sample size of 529 participants in the HFpEF study suggests there is still some distance to go before guidelines can be updated. The SELECT study’s transition to more complex clinical scenarios is the issue that attendees need to focus on most. The SELECT inclusion criteria are relatively clear: age ≥45 years, BMI ≥27, and a history of cardiovascular disease. However, the composition of real-world patients is far more complex.Obese patients with heart failure with preserved ejection fraction (HFpEF) may respond to GLP-1 in a manner entirely different from those with heart failure with reduced ejection fraction (HFrEF). The STEP-GLP-1 study demonstrated that semaglutide improved symptom scores in obese patients with HFpEF, but
diabetic status influences the transferability of these benefits. Since the majority of participants in the SELECT study had diabetes, is there a difference in cardiovascular benefits among the non-diabetic subgroup? Do the mechanisms by which GLP-1 affects insulin resistance and vascular endothelial function manifest differently in non-diabetic populations? This determines whether GLP-1 can be prescribed to patients with simple obesity who do not have diabetes but are at elevated cardiovascular risk—and this represents the largest potential market.
Patients with chronic kidney disease represent another key target population. The FLOW study evaluated 3,537 participants. If the renal protective effects of GLP-1 are confirmed, this will directly impact prescribing practices for patients with CKD.Further analyses of the FLOW study were presented at the ESC Congress 2026; attendees should pay attention to changes in the rate of decline in glomerular filtration rate (GFR), the extent of improvement in proteinuria, and the association between these renal markers and cardiovascular events. The renal composite endpoint for semaglutide in patients with diabetic nephropathy, as assessed in the
| Patient Subgroup | Shift in Focus | Current Evidence | Key Takeaways from ESC 2026 |
| HFpEF with obesity | Symptom Improvement vs. Hard Endpoints | STEP-HFpEF Phase II | Larger Sample Size, Hard Endpoints |
| Obesity in Non-Diabetic Patients | Proportional cardiovascular benefit | SELECT Subgroup | Confirmatory Analysis |
| Stage 3–4 CKD | Renal protection + CV events | FLOW Phase III | Renal Function Stratification MACE |
| Elderly (>75 years) | Safety + Functional Preservation | Limited | Safety Subgroup |
Long-term adherence and rebound after discontinuation range from 40 to 65%, far lower than in clinical trials. If patients experience rapid weight regain after discontinuation, does the cardiovascular protective effect disappear as well? Or does GLP-1 induce some lasting changes in vascular endothelium and myocardial metabolism during treatment? This question determines the pricing model for GLP-1—whether it should be priced based on lifelong use or on the duration of efficacy.The most pressing issue in GLP-1 commercialization. Real-world data show that the 12-month retention rate for GLP-1 injections is approximately
shifts attendees’ focus to real-world issues: patients with comorbid heart failure, diabetes, kidney disease, and the elderly; long-term adherence; and risks following treatment discontinuation. For a specific patient—age 68, with HFpEF, stage 3 CKD, and obesity—should the drug be prescribed based on the overall data from the SELECT trial, even though this patient’s specific characteristics may fall on the borderline of the SELECT enrollment criteria?What attendees need to look for at the ESC Congress is precisely subgroup data for these borderline populations. GLP-1? The real dilemma cardiologists face in their daily prescribing decisions. A
If we set aside MACE, could other metabolic regulatory mechanisms (such as GIP, GCG, and amylin) also produce similar cardiovascular benefits? This question is the core driving force behind the development of the next generation of cardiovascular metabolic drugs.GLP-1 should not be portrayed as the story of a single blockbuster drug, but rather as evidence of a paradigm shift in the treatment of cardiovascular and metabolic diseases. The true significance of GLP-1 lies not in its own merits, but in the fact that it has demonstrated that metabolic intervention can provide cardiovascular protection, opening up new avenues for drug development across the entire field of cardiovascular and metabolic medicine. If GLP-1 can reduce
1.2 The impact of the ESC’s “Hot Lines” on a drug’s commercial value is often more direct than that of ordinary industry conferences
There is a fundamental difference between top clinical conferences in the cardiovascular field and ordinary industry conferences: the former present hard-endpoint data, while the latter often discuss pipeline progress and commercial expectations. Each year, the ESC’s “Hot Lines” session selects several of the most influential “Late-Breaking Science” studies for their first public release. The selection criteria for these studies are extremely rigorous—they must be prospective randomized controlled trials that change clinical practice.
The ripple effect of this release mechanism is that positive results from a Hot Lines study may be incorporated into discussions regarding guideline updates within months of publication, thereby altering physicians’ prescribing habits and payers’ reimbursement decisions.For pharmaceutical companies’ business development (BD) teams, this means that valuation benchmarks for assets can shift rapidly; for investors, it means that capital expectations must be adjusted within a short timeframe. However, it is important to remain clear-headed: while the impact of Hot Lines is significant, it should not be exaggerated as “decisive”—a more accurate assessment is that it alters the market’s perception of the level of evidence, which in turn influences subsequent indication planning and serves as leverage in pricing negotiations.
| Year/Conference | Hot Lines Breakthrough Studies | Drugs/Therapeutic Areas Involved | Impact on Commercial Value |
| ESC 2023 | First results from the SELECT trial | Semaglutide (GLP-1) | 20% reduction in MACE, directly leading to the FDA’s approval of Wegovy for cardiovascular indications in March 2024; GLP-1 was repositioned from a weight-loss drug to a cardiovascular prevention drug |
| ESC 2023 | STEP-HFpEF | Semaglutide (GLP-1) | Demonstrated symptom improvement with GLP-1 in the HFpEF population, expanding the scope of potential heart failure indications |
| ESC 2024 | FLOW Trial | Semaglutide (GLP-1) | Positive renal composite endpoint paves the way for a CKD indication, further expanding the systemic value of GLP-1 |
| ESC 2025 | Multiple GLP-1 extension analyses | Semaglutide/tirzopentide | Data from complex patient subgroups influence guideline recommendations and reimbursement decisions |
| ESC 2026 (Expected) | Long-term GLP-1 Outcomes + Early Signals for Nucleic Acid Drugs | GLP-1/AOC/Small Nucleic Acids | May Simultaneously Validate Long-Term CV Benefits of GLP-1 and Early Clinical Signals for Cross-Tissue Delivery of Nucleic Acid Drugs |
A pattern evident from this table is that between the release of the ESC’s SELECT trial in 2023 and FDA approval for cardiovascular indications in 2024, less than five months elapsed. This pace implies that pharmaceutical companies treating the ESC merely as an academic conference will miss the most critical market window. The impact of “Hot Lines” on a drug’s commercial value follows a chain of transmission: “release—guidelines—prescription—reimbursement.”
The GLP-1 narrative of 2026 has already concluded its first chapter—“Is it effective?”—and now the second chapter must address “How long do the effects last, and how broad a patient population can it cover?”At the same time, if any early clinical signals related to cardiovascular outcomes for AOCs or small nucleic acid drugs are released during satellite meetings or workshops surrounding the ESC, the impact on industry expectations could be no less significant than a positive “Hard Line”—because this would represent the first clinical milestone marking the transition of nucleic acid drug platforms from proof of concept to cardiovascular indications. The reason Hot Lines warrant attention is that,
for attending pharmaceutical teams, the SELECT extension analysis—which shows that cardiovascular benefits diminish in specific subgroups—means that positioning strategies for competitors targeting the same pathway will need to be adjusted; the value of the Hot Lines lies not in hearing the results themselves, but in immediately assessing their impact on their own pipelines and the competitive landscape. If the SURPASS-CVOT trial for telaprotein yields a non-inferiority conclusion, the market narrative for dual-target drugs will need to be rewritten.These assessments must be made on-site, rather than waiting until returning to the office to read news summaries.
Action Recommendation: Before attending the ESC Congress 2026, list the 3–5 Hot Lines studies most relevant to your pipeline and prepare a rapid assessment framework for each study covering three scenarios: “positive,” “negative,” and “non-inferiority.” During the conference, focus on documenting the study’s primary endpoints, subgroup analyses, and safety updates; within 48 hours after the conference, complete a revised assessment of asset valuation and the competitive landscape.
The Hot Lines session is the most valuable part of the ESC Congress. The landmark clinical trials presented in this session each year often directly reshape the recommendations in the European Society of Cardiology guidelines, thereby influencing global cardiovascular clinical practice. For pharmaceutical companies, the presentation of a study during Hot Lines signifies endorsement by a top-tier academic platform, which holds far greater value in regulatory communications, health insurance negotiations, and physician education than academic events organized independently by the company.
Within 6 to 12 months of publication, adjustments to recommendation levels directly impact national health insurance reimbursement policies.The third layer is the clinical practice effect: within 1 to 2 years, the prescription rate for new drugs will change significantly during this period. The influence of Hot Lines is transmitted through multiple layers. The first layer is the immediate effect: after data is announced at the conference, specialized and financial media report on it within 24 hours, affecting pharmaceutical companies’ stock prices and investor expectations. The second layer is the guideline effect: ESC guideline updates typically occur after the conference, and cardiologists’ response to the guideline recommendations usually takes
| Impact Levels | Time Window | Affected Parties | Key Actions |
| Immediate Effects | 24–72 hours after release | Stock Price/Investors | Rapid Analysis and Internal Communication |
| Guidance Effect | 6–12 months after the meeting | ESC Guidelines/Health Insurance | Assessing the Impact of Guideline Changes |
| Impact on Clinical Practice | 1–2 years | Prescribing Behavior | Formulation of Market Strategies |
| Regulatory Impact | 1–3 years | EMA/FDA Review | Preparing Regulatory Submissions |
The timing of data releases on Hot Lines directly impacts the pace of transactions. If key data for a drug candidate performs exceptionally well at the ESC, the licensor’s valuation expectations will rise shortly after the conference, and the window for in-licensing negotiations may narrow. Conversely, if competitor data falls short of expectations, it creates a window for reassessing the valuations of comparable assets. The BD team needs to establish a framework for interpreting the data well in advance of the conference. For the BD team,
negative or unexpected results also appear in Hot Lines. These results are equally valuable; they correct market expectations, deflate valuation bubbles, and allow truly competitive projects to emerge. For investors, a Hot Lines release that falls short of expectations is often more informative than one that exceeds them, as it reveals which areas face technical limitations or barriers to clinical translation.
An ESC Class I recommendation (i.e., where evidence or consensus indicates that a treatment is beneficial, useful, and effective) significantly increases the likelihood of reimbursement approval under national health insurance programs. Conversely, if a study fails to be adopted by the guidelines after being presented at Hot Lines, or receives only a Class IIb recommendation (where benefits may outweigh risks but more data is needed), its commercial prospects will be substantially impacted.The reason Hot Lines have a more direct impact on a drug’s commercial value than ordinary industry conferences is the ESC guidelines’ authoritative status in global cardiovascular clinical practice. If a study is included in the ESC guidelines as
The capital markets often react faster than clinical practice. IR teams need to provide investors with a balanced interpretation immediately after data release, emphasizing positive findings while also pointing out the data’s limitations and uncertainties. Data presented at Hot Lines is reflected in the stock prices of relevant pharmaceutical companies within hours of release. However, this immediate reaction is often an overreaction—investors focus solely on overall endpoint data while overlooking subgroup analyses, safety signals, or limitations in study design. Pharmaceutical companies’
The impact of major ESC study presentations on guidelines, physician perceptions, market expectations, and future indication strategies is gradual and does not materialize immediately after the conference. Guideline updates typically take 6 to 12 months, as the ESC Guidelines Committee requires time to assess the quality of new evidence, conduct systematic reviews and meta-analyses, and coordinate with other guidelines.Changes in physician perception are even slower, typically taking 1 to 2 years, as the initial reaction—which often occurs within 24 to 72 hours—is frequently an overreaction. Cardiologists need time to gradually incorporate new evidence through continuing education, peer discussions, and clinical practice. Changes in capital expectations occur most rapidly, usually within
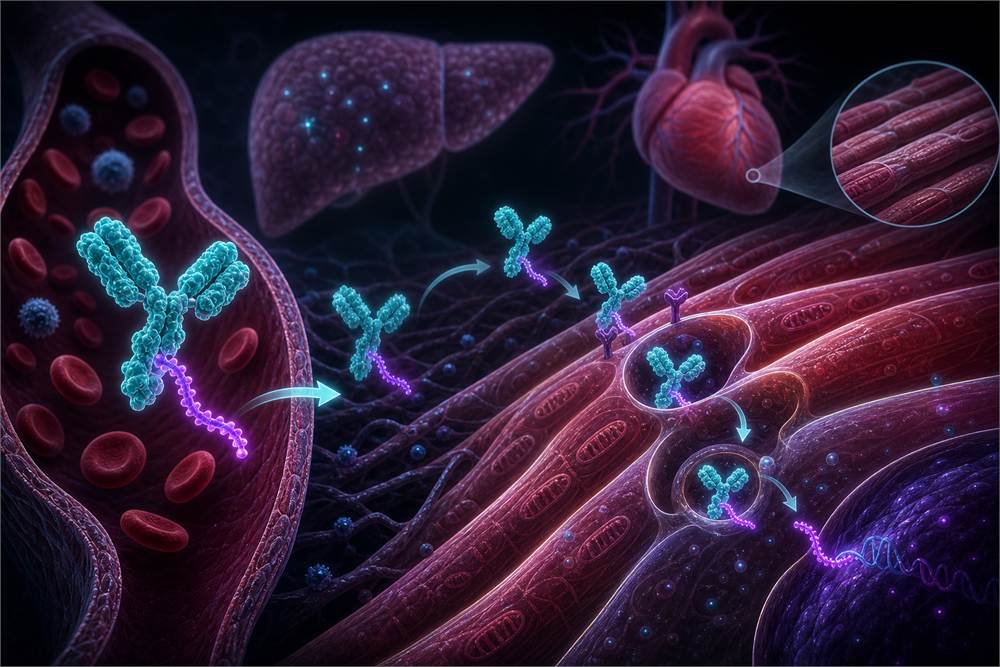
2. At the bio meeting 2026, the expansion of AOCs and small nucleic acids beyond the liver has raised a new question for the cardiovascular field: Can delivery platforms truly be reused across different tissues?
One of the hottest deals in the industry in 2026 was Novartis’ acquisition of AOC (antibody-oligonucleotide conjugate) for approximately $12 billion. The principle behind AOC is to use antibodies to precisely deliver small nucleic acids (siRNA/PMO) to extrahepatic tissues, including skeletal muscle, cardiac muscle, and even the central nervous system.This breaks the previous limitation that nucleic acid drugs were confined to “liver targeting,” reopening the space for targets in cardiovascular diseases, rare muscle disorders, and neurodegenerative diseases. The acquisition of Avidity Biosciences. The core asset of this deal is a technology platform itself, rather than a specific product,
but when viewed in the context of delivery platforms expanding from the liver to cardiac muscle, can it maintain sufficient delivery efficiency, safety, and tissue selectivity to support a development pathway for cardiovascular indications? If this question cannot be answered, the $12 billion acquisition price will remain merely a conceptual vision for the platform, unable to translate into product value. The real question is not “how hot AOC is,” but rather: against the backdrop of the ESC Congress 2026, the issue truly worth discussing is not “
From the perspective of industry signals, Novartis’ acquisition of AOC and the progress in small nucleic acids should be kept under observation. If nucleic acid drugs can be effectively delivered to myocardial tissue, a new branch will emerge in the development landscape of cardiovascular drugs. “Avidity” conveys three layers of meaning.First, competition in nucleic acid therapeutics has shifted from “who can develop them” to “where they can be delivered”; second, the technical barriers to extrahepatic delivery are sufficient to justify a $10 billion transaction valuation; third, large pharmaceutical companies are willing to pay a premium for platform technology, but this premium must ultimately be justified by clinical data and product launches. While attendees focus on cardiovascular data at the ESC Congress, they should also consider
Novartis’ acquisition of the AOC platform has already cleared the proof-of-concept hurdle. However, from a technical perspective, significant uncertainty remains regarding the maturity of the AOC platform.The Avidity deal should not be viewed simply as a routine M&A transaction. It marks a technological inflection point in the delivery of nucleic acid therapeutics—shifting from liver-targeted delivery to extrahepatic tissues. At least from the perspective of capital markets and the strategic priorities of major pharmaceutical companies, there is currently no clinical data to support whether the delivery efficiency observed in skeletal muscle for DMD can be transferred to cardiac muscle, vascular smooth muscle, or other cardiovascular-related tissues.When evaluating investment or collaboration opportunities in the AOC field, participants need to distinguish between platform value and the value of specific indications at this biotech conference Europe.
2.1 The commercial vision for small nucleic acids is shifting from “long-acting” to “where it can be delivered”
Small nucleic acid drugs (requiring administration only once every six months or even once a year). Inclisiran (Leqvio), currently the only approved siRNA drug for cardiovascular indications, achieves liver-targeted delivery via GalNAc conjugation and is used to lower LDL-C, requiring only two injections per year. For patients with chronic cardiovascular conditions requiring lifelong medication, this dosing frequency addresses a core pain point related to adherence. The underlying advantages of siRNA have always been clear:
However, long-acting effects are merely a result, not the entirety of their competitive edge. If all underlying competitive factors are shifting from “dosage frequency” to a comprehensive comparison across these five dimensions—“delivery systems, tissue distribution, target accessibility, immune response, and safety for repeated dosing”—the breakthrough in AOC technology has prompted an industry-wide reassessment precisely because it has, for the first time, expanded the delivery range of siRNA from the liver to muscle tissue, opening up an entirely new space for therapeutic targets.If siRNA can only reach the liver, then accessible targets are limited to genes expressed in the liver, and the competitive landscape will quickly become crowded.
| Delivery Systems | Target Tissue | Representative Drugs/Technologies | Dosage Frequency | Validated Indications | Technical Bottlenecks |
| GalNAc Conjugation | Liver | Inclisiran (Leqvio) | Once every 6 months | Hypercholesterolemia, HeFH | Liver-targeted only; limited target space |
| LNP (lipid nanoparticle) | Liver (primarily) | Patisiran (Onpattro) | Infusion every 3 weeks | Hereditary transthyretin amyloidosis | Risk of infusion reactions and hepatotoxicity; stringent storage and transport requirements |
| AOC (antibody-oligonucleotide conjugate) | Skeletal muscle, cardiac muscle, CNS (potential) | Delpacibart etedesiran (AOC 1001) | Intravenous infusion; frequency to be determined | DMD (clinical trial phase) | Delivery efficiency, tissue selectivity, immunogenicity, CMC complexity |
| EXG (exon skipping) + AOC | Skeletal muscle | AOC 1044 | To be determined | DMD exon 44 skipping (clinical stage) | Uniformity of muscle distribution, long-term safety |
| Direct central nervous system delivery (intrathecal) | Central Nervous System | Nusinersen (Spinraza) | Intrathecal injection every 4 months | Spinal Muscular Atrophy (SMA) | Technical barriers to invasive administration and intrathecal injection |
This table reveals a key fact: different delivery systems determine different target spaces. If AOC can achieve effective delivery into cardiac muscle tissue, it would open a previously closed door to cardiovascular indications. However, the final column in the table, “Technical Bottlenecks,” represents the true hurdle—is the delivery efficiency high enough? Is the tissue selectivity sufficient? Is there sufficient safety data for long-term repeated dosing? There are currently no definitive answers to these questions.GalNAc and LNP restrict nucleic acid drugs to the liver, whereas
from a commercial perspective, the value anchor for small nucleic acid drugs is shifting.In the past, the market valued AOCs for their ability to stably deliver siRNA to cardiac muscle; with that capability, siRNA drugs for cardiovascular indications were no longer just a theoretical possibility but a realistic option in the pipeline. The premium for siRNA primarily stemmed from the convenience of “ultra-long-acting” dosing; now, the market is beginning to pay for “where it can be delivered.” Novartis’s $12 billion acquisition of Avidity was, in essence, a purchase of “tissue options for extrahepatic delivery.” If
The core appeal of small nucleic acid drugs lies in their long-acting nature. Administered via subcutaneous injection, inclisiran maintains its LDL-C-lowering effect with just one dose every six months. This breakthrough in dosing frequency relies on the RNA interference mechanism—once siRNA enters a cell and is loaded into the RISC complex, its mRNA silencing effect can last from weeks to months. However, long-acting properties are merely a result, not a barrier.The true technical hurdle lies in the delivery system. siRNA drugs such as
the vast majority of currently approved small nucleic acid drugs rely on liver-targeted delivery. GalNAc-conjugation technology enables liver-specific uptake by binding to the ASGPR receptor.The delivery efficiency of this system is already well-established—siRNA is conjugated to N-acetylgalactosamine and utilizes Alnylam’s Onpattro, Givlaari, Oxlumo, and Leqvio, all of which are based on this platform. However, there are currently no equivalent targeted delivery solutions for tissues outside the liver. This is the crux of the “ex-liver” challenge.
| Delivery Platforms | Target Tissues | Representative Products | Maturity | Key Challenges |
| GalNAc Conjugation | Liver | Inclisiran | On the market | Liver-only |
| AOC antibody-drug conjugate | Muscle/Myocardium | AOC 1001 (DMD) | Phase I/II Clinical Trials | Conjugation consistency; DAR distribution |
| LNP (lipid nanoparticles) | Liver/Local | patisiran | Marketed | Systemic administration; poor extrahepatic distribution |
| Exosomes | Can be engineered | Early clinical | R&D stage | Yield; characterization |
AOC (Avidity Biosciences’ AOC 1001 demonstrated dose-dependent dystrophin restoration in a Phase 1/2 clinical trial in DMD patients, marking the first clinical data to validate the feasibility of the AOC platform in humans.Antibody-oligonucleotide conjugates (AOCs) are currently the most promising extrahepatic delivery strategy. They combine the tissue-specific targeting of monoclonal antibodies with the gene-silencing effects of oligonucleotides; the antibody directs the drug to the surface of specific cells, while the oligonucleotide exerts its effect within the cell.
From the perspective of cardiovascular drug development, the TGF-beta pathway—previously considered an undruggable target due to the difficulty of achieving selective inhibition with small molecules and the difficulty of large molecules entering cells—now holds new promise. The significance of AOCs, or AOCs combined with small nucleic acids, in delivering therapeutics beyond the liver lies in opening up an entirely new space of therapeutic targets.For many key pathological proteins in cardiovascular diseases—such as troponin in cardiomyocytes, calcium channel subunits in vascular smooth muscle, or siRNAs associated with myocardial fibrosis—targeted delivery to the myocardium or vascular smooth muscle can be achieved, allowing these targets to re-enter the development pipeline.
Long-acting properties are merely a result; the underlying competitive advantages lie in the delivery system, tissue distribution, target accessibility, immune response, and safety for repeated dosing.These five dimensions constitute a comprehensive framework for evaluating the value of a small nucleic acid platform. The delivery system determines whether the drug can reach the target tissue; tissue distribution determines the concentration ratio of the drug in target versus non-target tissues; target accessibility determines whether the drug can effectively reach the site of action after reaching the target cell; the immune response determines the safety window for repeated dosing; and the safety of repeated dosing determines the feasibility of long-term treatment regimens.
From an industry competition perspective, the small nucleic acid field is shifting from a focus on who has liver-targeted products to who can deliver to extrahepatic tissues.AOC still holds a significant first-mover advantage with its GalNAc platform, but Ionis, Arrowhead, and siRNA have demonstrated potential in preclinical studies; Avidity’s ASO already has an approved product (Spinraza); and Arrowhead’s muscle-targeting Avidity is advancing its own extrahepatic delivery strategies.Ionis’s central nervous system-targeted platform, Alnylam, already has human data in DMD. The key turning point in this competition will be which platform is the first to demonstrate clinical signals in cardiovascular indications.
Long-acting formulation is merely an outcome; the underlying competitive factors are the delivery system, tissue distribution, target accessibility, immune response, and safety for repeated dosing. Among these five dimensions, the delivery system is the most fundamental, as it determines whether the drug can reach the target tissue.Although AOC has demonstrated proof of concept for skeletal muscle targeting via antibody-mediated delivery, the actual delivery efficiency—that is, the percentage of the administered dose that reaches the target tissue—remains relatively low. In contrast, GalNAc-conjugation technology has achieved high delivery efficiency for liver targeting, with hepatocyte uptake rates reaching over 50% of the dose. However, extrahepatic tissues lack similarly mature delivery systems.This can be indirectly inferred from early clinical data on DMD.
Immune responses are a key variable in the safety of repeated dosing. Oligonucleotides inherently possess some immunostimulatory activity—in particular, those containing specific sequence motifs (such as the CpG motif) can activate the innate immune system via Toll-like receptors.Although chemical modifications (such as 2′-O-methylation, 2′-fluorination, and thio-phosphate backbones) can reduce immunostimulation, it is difficult to eliminate it entirely. The antibody component in AOCs may also trigger ADA. The cumulative effect of immune responses following repeated dosing is a safety concern that requires close monitoring for the AOC platform in the long-term treatment of chronic cardiovascular diseases.
2.1.1 For cardiovascular drug developers, extrahepatic delivery offers practical value and serves as a gateway to new therapeutic targets
When viewed collectively, the technological breakthroughs in AOC reveal an interesting intersection: many disease-causing genes in cardiovascular disease are expressed in cardiac muscle and vascular smooth muscle, but these tissues were previously “inaccessible” to GalNAc—which could only reach the liver—and LNP, which is also primarily distributed in the liver. This means that any therapeutic strategy requiring direct regulation of gene expression in cardiac muscle has historically lacked a viable delivery method.For siRNA, these tissues were “unreachable.”
Silencing of genes associated with cardiac hypertrophy (such as MYBPC3 and MYH7), regulation of ion channels linked to arrhythmias, and local inhibition of vascular inflammatory factors. These targets are either difficult to develop into drugs using traditional small molecules and antibody therapies, or systemic administration results in unacceptable side effects. If AOC can achieve effective delivery to the myocardium, cardiovascular drug developers will gain a ticket to a new space of therapeutic targets.Specific indications may include: Tissue-specific siRNA silencing could theoretically resolve this contradiction.
| Potential Cardiovascular Targets | Tissue of Target | Accessibility of Traditional Drugs | Potential Advantages of AOC/siRNA | Current stage of evidence |
| MYBPC3 (myosin-binding protein C) | Myocardium | Small molecules are difficult to regulate precisely, and gene therapy is still in its early stages | siRNA can silence mutant transcripts while preserving wild-type function | Preclinical studies |
| PCSK9 (proprotein convertase subtilisin/kaleasin 9) | Liver (GalNAc-siRNA already available) | Antibody drugs and siRNA (Inclisiran) are already available | There are precedents for liver delivery; AOC can be extended to local delivery to the vascular wall | Approved (liver delivery) |
| Connexin-43 (gap junction protein 43) | Cardiac muscle + vascular smooth muscle | Safety concerns surrounding small-molecule modulators are significant | Tissue-specific silencing may reduce arrhythmia-related side effects | Preclinical |
| Inflammatory factors (e.g., TNF-α, IL-6) | Vascular wall + myocardium | Systemic administration carries significant side effects, and cardiovascular benefits are uncertain | Local delivery may achieve anti-inflammatory effects without suppressing systemic immunity | Proof-of-concept stage |
| DMPK (Dystrophin Kinase) | Skeletal muscle + cardiac muscle | No effective targeted drugs | AOC delivers siRNA to silence toxic RNA; clinical data for DMD are available for reference | AOC 1001 is currently in clinical trials |
Most of the targets listed in this table are currently in the preclinical or proof-of-concept stages, and the technical pathway for migrating the AOC platform to cardiac muscle tissue is feasible. It will still be some time before clinical signals emerge at the ESC Congress 2026. However, the areas attendees should focus on are clear: Which indications will show clinical signals first? Which endpoints best demonstrate the platform’s value?Based on Avidity’s DMD clinical trials, exon skipping in muscle tissue and functional improvement metrics (such as the Polaris Dynamic Assessment—NSAA) are currently the first types of endpoints being validated. If these metrics remain consistently positive in the DMD trials, observers in the cardiovascular field will have reason to believe that
What requires caution—and what attendees should be asking—is: Were any animal model data on myocardial delivery or early clinical signals presented at the satellite meetings surrounding the ESC? If so, even data at the proof-of-concept level would be sufficient to alter the industry’s expectations regarding the timeline for the AOC platform’s application in the cardiovascular field. Data on AOC delivery efficiency in skeletal muscle cannot be directly extrapolated to the myocardium.Clinical validation in DMD does not mean that the approach can be directly applied to cardiovascular indications. Skeletal muscle and cardiac muscle differ significantly in terms of blood supply, cellular structure, delivery barriers, and immune environment.
The significance of extrahepatic delivery for cardiovascular drug developers must be understood from two perspectives: target accessibility and clinical endpoints. Cardiomyocytes are terminally differentiated cells; drugs must traverse the capillary wall, the interstitium, and the cardiomyocyte membrane to reach their site of action.The distribution of traditional small-molecule drugs in myocardial tissue is limited by P-glycoprotein efflux and degradation by metabolic enzymes. AOCs, through antibody-mediated targeted delivery, can theoretically bypass these barriers; however, actual delivery efficiency, the proportion of distribution in myocardial tissue, and off-target distribution remain unknown.
From an indication perspective, the cardiovascular field is most likely to see clinical signals emerge first in ATTR-CM (as demonstrated by patisiran), myocardial fibrosis (targeting TGF-beta or LOXL2), and arrhythmias (targeting specific ion channel subunits). These indications are characterized by relatively well-defined pathological mechanisms, intracellular targets, and limited existing treatment options.AOC directions include: hereditary cardiomyopathies such as transthyretin amyloidosis
| Potential AOC Indications | Target Areas | Limitations of Current Treatments | AOC’s Differentiated Value |
| ATTR cardiac amyloidosis | TTR (myocardial deposition) | Tafamidis only delays progression; liver transplantation is limited | Directly silences TTR locally in the myocardium |
| Myocardial fibrosis | TGF-beta/LOXL2 | No approved anti-fibrotic drugs | Targeting myocardial fibroblasts |
| Arrhythmia | Ion channel subunits | Existing drugs lack selectivity | Cell-type-specific silencing |
| Cardiomyopathy | MEF2/HDAC pathway | No targeted therapies | Epigenetic regulation |
While attendees are focusing on the therapeutic potential of AOC, they must also be acutely aware of the risks associated with platform translation. Moving from DMD to cardiovascular applications involves more than simply switching antibody targets. Receptor expression levels, endocytosis efficiency, endosomal escape rates, and the intracellular stability of oligonucleotides vary across different tissues.There are currently no clinical data to support whether AOCs targeting skeletal muscle in DMD can be proportionally transferred to cardiac muscle tissue. Attendees need to monitor whether the AOC platform demonstrates evidence of tissue selectivity—that is, whether drug concentrations in the target tissue are significantly higher than in non-target tissues.
Another key issue is repeat dosing. Cardiovascular diseases typically require lifelong treatment, and concerns regarding AOCs in DMD have already begun to emerge in early-stage clinical trials. Clinical trial designs for cardiovascular indications must incorporate ADA monitoring and immunogenicity management protocols from the outset.Immunogenicity—particularly the production of anti-drug antibodies (ADAs)—may affect efficacy and safety following repeated dosing. If patients develop neutralizing antibodies after multiple doses, this will not only reduce therapeutic efficacy but may also trigger immune-related adverse events. This issue in
has a natural connection to the ESC’s cardiovascular clinical scenarios in areas such as the myocardium, skeletal muscle, and the nervous system. The myocardium is the most direct target tissue for cardiovascular drugs; although skeletal muscle is not a cardiovascular target tissue, the validation of AOC delivery in skeletal muscle provides a technical reference for myocardial delivery;the central nervous system is related to cardiovascular autonomic regulation, particularly the neural regulation of arrhythmias and sudden cardiac death. Attendees should observe which indications will show clinical signals first and which endpoints best demonstrate the platform’s value.
Which indications will show clinical signals first? From the perspective of technical feasibility, effective delivery is easier to achieve in tissues that highly express the target receptor. Transthyretin amyloid cardiomyopathy (ATTR-CM) is a potential direction—TTR is primarily synthesized in the liver, but its deposition in the myocardium is the pathogenic mechanism.If patisiran exhibits greater tissue specificity, the feasibility of this strategy depends on whether TTR mRNA expression levels in the myocardium are sufficient to serve as a target for the AOC. An AOC capable of directly silencing TTR mRNA expressed locally in the myocardium (where there is also localized TTR expression) is often more effective than one targeting the liver.
Which endpoints best demonstrate the platform’s value? For cardiovascular AOCs, the most direct endpoint is the efficiency of target protein silencing in myocardial tissue; however, this requires verification via myocardial biopsy, which is highly impractical in a clinical setting.Surrogate endpoints include changes in target protein concentrations in the blood, improvements in cardiac function parameters (such as LVEF and LVESD), and changes in cardiac biomarkers (such as NT-proBNP and troponin). However, the quantitative relationship between these surrogate endpoints and AOC delivery efficiency has not yet been established and needs to be gradually clarified through early pharmacokinetic-pharmacodynamic (PK-PD) studies.
2.2 The Greater the Interest in AOCs, the More Critical It Is to Ensure CMC and Analytical Methods Can Keep Pace
When a $12 billion acquisition brings CMC (Chemistry, Manufacturing, and Control) and analytical methods into the picture.AOC is not simply a matter of mixing antibodies and oligonucleotides. As a conjugated molecule, it involves multiple stages—including antibody production, oligonucleotide synthesis, conjugation reactions, purification, and formulation—each with its own unique technical barriers and quality control requirements. With AOC thrust into the spotlight, rational conference attendees should instead turn their attention to what lies beyond the spotlight:
As the industry shifts from the hype surrounding platform transactions to the commercialization of products, AOCs—as conjugates—add antibody-related manufacturing complexities on top of these existing challenges.CMC is an unavoidable hurdle. Currently, the overall CMC challenges for oligonucleotide drugs are already quite complex—according to a review published in *Medicinal Chemistry Research* in 2026, despite nearly three decades having passed since the approval of the first oligonucleotide drug, Fomivirsen, this class of drugs still faces unique CMC challenges in API development.
| CMC Dimensions | Small-Molecule Drugs | Conventional Antibody Drugs | Oligonucleotide Drugs | AOC (Conjugates) |
| Molecular Structure Confirmation | Relatively simple; NMR/MS is sufficient | Requires a full set of characterization (peptide maps, glycan profiles, etc.) | Sequence confirmation and identification of modifying groups | Antibody + oligonucleotide dual characterization + confirmation of conjugation sites |
| Impurity profile control | Organic impurities + inorganic impurities | Host cell proteins, aggregates, fragments | Sequence-related impurities (shortmers/longmers), sequence deletions | All of the above + free oligonucleotides + free antibodies + conjugation byproducts |
| Conjugation Consistency | Not applicable | Not applicable | Not applicable | Drug-to-antibody ratio (DAR) distribution, uniformity of conjugation sites |
| Batch-to-batch consistency | Mature process, easy to control | Cell line stability + process control | Synthesis yield + batch-to-batch variation in purification | Triple sources of variation: antibody batch, oligonucleotide batch, and coupling efficiency |
| Stability | Relatively mature | Cold chain requirements, aggregate monitoring | Sensitive to nuclease degradation and freeze-thaw cycles | Antibody stability + oligonucleotide stability + conjugation bond stability |
| Scale-up production | Chemical Synthesis, Scale-Up, and Maturation | Bioreactor Scale-Up | Scale-up of solid-phase synthesis is limited; liquid-phase synthesis is under development | Separate scale-up of antibodies and oligonucleotides + scale-up of the conjugation process |
This comparison table illustrates a core issue: the complexity of AOC’s CMC is multiplicative, not simply additive. Batch-to-batch variability in antibodies, sequence purity of oligonucleotides, and the efficiency of the coupling reaction—the sources of variability in these three factors combine, making it far more difficult to control batch-to-batch consistency than with a single molecular type.For quality management teams, this means that the establishment of release criteria must simultaneously cover antibody-related impurities (aggregates, fragments, host cell proteins), oligonucleotide-related impurities (shortmers, longmers, sequence deletions), and coupling-specific impurities (free oligonucleotides, free antibodies, coupling byproducts).
In 2024, the EMA published a draft guideline on the development and manufacture of oligonucleotide medicinal products, which explicitly mentions control requirements for non-oligonucleotide impurities (process reagents and their byproducts, residual solvents, elemental impurities, and potential mutagenic impurities).Although this guidance is not directly aimed at AOC, its requirements for analytical methods related to oligonucleotide impurities—including the use of LC-MS for sequence confirmation and impurity identification—provide a foundational framework for the analytical characterization of AOC. Attendees interested in the AOC platform should be aware of these regulatory trends, as they directly impact the platform’s path from clinical trials to market approval.
AOC still has a long way to go from the current transaction frenzy to actual product commercialization. The $12 billion acquisition price reflects market expectations for extrahepatic delivery platforms, but these expectations ultimately need to be fulfilled through clinical data, CMC maturity, and regulatory engagement. When assessing the value of the AOC platform, attendees should consider the maturity of CMC and analytical methods as one of the core evaluation criteria, rather than focusing solely on transaction values and clinical proof-of-concept data.
Action Recommendation: Pay attention to whether there are sessions on the development strategies and commercialization timelines for the AOC platform for nucleic acid therapeutics on the ESC program. Look for workshops or satellite sessions on CMC and analytical methods. At the same time, track updates to EMA and FDA guidelines on oligonucleotide drugs, as changes to these regulatory documents will directly impact
AOC is not merely a conceptual combination of an antibody and an oligonucleotide. Its CMC complexity far exceeds that of either component alone.The antibody component must retain its original binding activity and stability; the oligonucleotide component must maintain its gene-silencing activity after conjugation; and the linker must be stable in circulation and cleavable within target cells. The quality control standards for these three components, when combined, form an entirely new analytical characterization system.
Conjugation uniformity is the first technical hurdle. The number of oligonucleotides attached to each antibody molecule (AOC) requires controlling the DAR distribution within a narrow range. However, the molecular weight of oligonucleotides is much greater than that of small-molecule toxins (approximately 13–15 kDa vs. 1 kDa), which means greater steric hindrance at each conjugation site and more difficult kinetic control of the conjugation reaction.The DAR (Drug-to-Antibody Ratio) directly affects efficacy and safety. An excessively high DAR can lead to antibody aggregation and off-target toxicity; conversely, an excessively low DAR reduces delivery efficiency. Similar to ADCs,
| CMC Aspects | AOC-Specific Challenges | Comparison with ADCs | Comparison with Naked Oligonucleotides |
| Conjugation Consistency (DAR) | Large oligonucleotide molecular weight; steric hindrance | Small-molecule toxins; DAR controls maturation | No coupling step |
| Impurity profile | Oligonucleotide-related impurities (shortmers/longmers) | Small-molecule-related impurities | Impurity profiling is relatively straightforward |
| Free oligonucleotides | Unconjugated free oligonucleotides must be controlled | Free toxins must be controlled | Are the final product themselves |
| Stability | Dual stability requirements for antibodies and oligonucleotides | Antibody + small molecule | Oligonucleotide stability on its own |
| Analytical Methods | New conjugation characterization methods are required | Established methods such as HIC-HPLC | Ion-exchange HPLC |
Oligonucleotide-related impurities include shortmers (fragments shorter than the target sequence) and longmers (fragments longer than the target sequence), as well as various incompletely modified byproducts.While there is already a well-established set of analytical and release standards for these impurities in free oligonucleotide drugs, in AOCs they may exist in free form or be conjugated to antibodies, significantly increasing the complexity of analysis and characterization. Unique CMC challenges distinguishing AOCs from ADCs. The solid-phase synthesis of oligonucleotides generates
AOC production involves four major steps: antibody expression and purification, oligonucleotide synthesis and purification, the conjugation reaction, and final formulation. Batch-to-batch variability in each step accumulates in the final product.In early clinical stages, small-batch manual operations may mask these issues; however, once commercial-scale production is reached, even minor process deviations can lead to shifts in the DAR distribution, an increased proportion of free oligonucleotides, or increased antibody aggregation. Establishing robust process characterization and design space is essential for AOCs to transition from clinical to commercial production. Batch-to-batch consistency is another critical issue in scale-up.
Stability studies also require redesign. The stability of AOCs is influenced by both the antibody and the oligonucleotide: the antibody may undergo deamination or oxidative degradation; the oligonucleotide may undergo phosphodiester bond hydrolysis or depurination; and the conjugation bond may slowly cleave during storage.This means that stability study protocols for AOCs cannot simply be derived from existing templates for antibodies or oligonucleotides; rather, dedicated stability testing methods tailored to the conjugated product must be developed.
The introduction of the QbD (Quality by Design) philosophy is crucial. QbD requires defining Critical Quality Attributes (CQAs) during the product design phase, establishing mathematical relationships between Critical Process Parameters (CPPs) and CQAs, and ensuring process robustness through design space analysis. For AOCs, CQAs involve Design of Experiments (DOE) studies, which require significant investments of both time and resources.On the path from market buzz to product commercialization for AOCs, key factors include DAR distribution, free oligonucleotide proportion, aggregate content, potency assay values, and stability metrics. CPPs, meanwhile, encompass the pH, temperature, molar ratio, and reaction time of the coupling reaction, as well as the cleavage conditions during purification steps. Establishing the CQA-CPP relationship requires a substantial amount of
| QbD elements | Specific content in the AOC | Maturity | Impact on Commercialization |
| CQA definition | DAR; Free Oligonucleotides; Aggregates; Efficacy | Partially Established | Basis for Release Criteria |
| CPP Identification | Coupled pH/Temperature/Molar Ratio; Purification Conditions | Early stages | Prerequisites for process lock-in |
| Design Space | Acceptable CPP Range | Not established | Process Change Management |
| Control Strategy | Process Control + Release Inspection | Basic Level | Batch Consistency Assurance |
The lead time for oligonucleotide raw material supply is also 18 to 24 months. If an AOC product enters late-stage clinical trials or the commercialization phase, the security of the oligonucleotide raw material supply chain will become an easily underestimated bottleneck in AOC commercialization.Solid-phase synthesis of high-purity oligonucleotides requires specialized phosphoramidite monomers, solid-phase supports, and synthesis instruments. The number of global suppliers capable of providing GMP-grade oligonucleotide raw materials is limited, and the capacity expansion cycle typically requires the CMC team’s close attention.
Process transfer from pilot scale (milligrams) to GMP commercial scale (grams to kilograms). AOC is not merely a conceptual combination of an antibody and an oligonucleotide; it involves coupling consistency, impurity profiles, oligonucleotide-related impurities, batch-to-batch consistency, stability, and scale-up production. Each of these aspects has its own technical barriers and quality standards.Conjugation consistency determines whether the number of oligonucleotides attached to each antibody molecule is uniform; the impurity profile covers antibody-related impurities, oligonucleotide-related impurities, and byproducts of the conjugation reaction; batch-to-batch consistency must be ensured through process validation; stability studies must cover the three degradation pathways of the antibody, oligonucleotide, and conjugation bond; and scale-up production requires progression from laboratory scale (
| AOC CMC Dimensions | Technical Barriers | Current Maturity | Key Issues to Be Resolved Before Commercialization |
| Coupling Consistency | DAR Distribution Control; Coupling Site Selectivity | Moderate | Narrow DAR distribution process; confirmation of coupling sites |
| Impurity profile | Free oligonucleotides; shortmers/longmers; coupling byproducts | Partially established | Analysis method for conjugated impurities |
| Batch-to-batch consistency | Process parameter control; raw material consistency | Early | Process validation; design space establishment |
| Stability | Triple degradation of antibodies, oligonucleotides, and linkers | Basic level | Development of Stability Assay Methods |
| Scale-up production | Scaling up of the coupling reaction; scaling up of purification | Laboratory Scale | Process transfer; GMP facility validation |
Readers will thus see that such platforms still have a long way to go from transaction hype to product commercialization. Since it will take Novartis 2 to 3 years to support an IND filing for cardiovascular indications, the discount rate for the platform premium should be adjusted upward accordingly. The premium reflecting the AOC platform’s cross-indication potential is a question worthy of deep consideration by the BD team: how much of the transaction value for Avidity reflects the valuation of the DMD indication, and how much reflects the platform’s

3. At the bio meeting 2026, GLP-1, AOC, and new cardiovascular drugs all point to one change: clinical narratives must withstand scrutiny from manufacturing and quality systems
The competition for innovative cardiovascular drugs in 2026 is shifting from “whether the mechanism is novel” to “whether the evidence is robust enough and whether the product can be delivered consistently.”GLP-1’s cardiovascular outcome data must withstand scrutiny regarding long-term safety and complex patient populations; AOC’s extrahepatic delivery platform must withstand scrutiny regarding the findings from the 2026 ESC Congress and Novartis’ acquisition of CMC and analytical characterization capabilities; and traditional small-molecule and large-molecule cardiovascular drugs must withstand scrutiny regarding their differentiated value.Although these three categories of drugs differ in form, the challenges they face are fundamentally the same: no matter how compelling the clinical narrative, if manufacturing and quality systems cannot keep pace, the product cannot be brought to market. When reviewing Avidity’s trending news together, a common thread emerges:
For attendees, the significance of this shift lies in the need to simultaneously consider—while reviewing data at the ESC—where the subsequent development of dosage forms, process scale-up, analytical methods, and the supply chain might face pressure if the data holds true. This cross-disciplinary perspective, spanning from clinical trials to manufacturing, is an increasingly rare yet valuable capability in cardiovascular drug pipeline development in 2026.
The competition for innovative cardiovascular drugs in 2026 is shifting from the novelty of the mechanism of action to the robustness of the evidence and the ability to deliver products reliably. This assessment can be understood on three levels. First, GLP-1 has already demonstrated cardiovascular benefits; the next phase of competition will hinge on who can provide more robust endpoint data across a broader patient population.Second, AOC and small nucleic acid platforms need to demonstrate tissue selectivity and safety in cardiovascular indications, rather than merely providing proof of concept in DMD or rare diseases. Third, all new cardiovascular drugs must address a manufacturing-level question: Can the product maintain batch-to-batch consistency during large-scale production and be delivered reliably through the global supply chain?
3.1 The GLP-1 competition is entering the second half; formulation, production capacity, patient adherence, and differentiated endpoints will become increasingly important
The first half of the race focused on efficacy data; the second half will center on formulation convenience, production capacity, patient adherence, and the persuasiveness of differentiated endpoints.The competitive landscape for SELECT and GLP-1 receptor agonists is entering a new phase. The early phase of competition centered on “which drug delivers better weight loss”—semaglutide and tirzopotide engaged in a data race spanning several years, focusing on the magnitude of weight loss. However, once the SURPASS-CVOT study brought cardiovascular outcome data to the forefront, the dimensions of competition began to diverge:
| Competitive Dimensions | First Half (2020–2024) | Second Half (Projected 2025–2028) | Challenges for CMC and Manufacturing |
| Key Metrics | Weight Loss (STEP Series) | Cardiovascular hard endpoints + long-term safety + complex patient subgroups | Increased demand for long-term stability data; stricter requirements for batch consistency |
| Formulation Competition | Weekly formulations predominate (semaglutide/terliprotide injections) | Oral formulations + monthly formulations + ultra-long-acting formulations | Processing challenges to improve oral bioavailability; release profile control for monthly formulations |
| Competition in production capacity | Expansion of API synthesis capacity | Formulation filling capacity + dosing device (pen-style injector) production capacity | Precision manufacturing and quality consistency of delivery devices; scaling up cold-chain logistics |
| Competition in Adherence | Dosage Frequency (Once Weekly vs. Once Daily) | Administration experience (pain, injection volume) + oral convenience | Formulation optimization (buffer system, osmolarity, viscosity) |
| Differentiated endpoints | HbA1c + Body Weight | MACE + heart failure + kidney disease + NASH + sleep apnea | Dosage forms and administration regimens may vary across different indications, increasing SKU complexity |
| Multi-target design | GLP-1 single-target vs. GLP-1/GIP dual-target | GLP-1/GIP/GCG triple-target + other combinations | Longer/more complex peptide sequences make it more difficult to control synthesis purity and folding accuracy |
The last two columns of this table reveal the true barriers to entry in the latter half of the GLP-1 competition. Taking oral formulations as an example, Novo Nordisk’s oral semaglutide (Rybelsus) utilizes SNAC absorption-enhancing technology, yet its oral bioavailability is only about 1%. This implies extremely high requirements for API purity and batch-to-batch consistency—even the slightest variation in impurities could affect absorption rates.Another example is the development of monthly or ultra-long-acting formulations. If the dosing frequency is to be reduced from once a week to once a month or even longer, controlling the drug release profile becomes a core process challenge, and the batch-to-batch stability of the release profile directly affects the consistency of clinical efficacy.
The complexity of quality control arising from combination or multi-target designs should also not be overlooked. While GLP-1/GIP dual-target molecules are currently in development, some companies are already working on GLP-1/GIP/GCG triple-target molecules.Multi-target peptides typically have longer sequences, which may increase the proportion of missing or truncated sequences during synthesis and make purification more difficult. At the same time, longer peptide chains may affect folding and conformational stability, placing higher demands on analytical characterization methods. From a CDMO perspective, this means that suppliers with high-quality peptide synthesis and purification capabilities will become increasingly scarce and valuable. Telporpeptide is
Pressure on production capacity is even more immediate. For CDMOs and supply chain companies, competition in the second half of the GLP-1 era means that the ability to scale up filling capacity, administer device manufacturing, and cold-chain logistics is becoming a competitive factor just as important as clinical data.Both Novo Nordisk and Eli Lilly are significantly expanding their production capacity, but whether this expansion can keep pace with the growth in demand driven by indication expansion remains an open question. Semaglutide faced a global shortage in 2022–2023 due to insufficient production capacity, and the patient waiting list for Wegovy at one point stretched for months. Although
Action Recommendations: After reviewing a pharmaceutical company’s R&D and GLP-1 data, three questions should be immediately raised: ① Can the current dosage form support the administration regimen for the new indication? ② Does the production capacity plan cover the increased demand resulting from the expanded indication? ③ Is the data on differentiated endpoints sufficient to support pricing negotiations, or is additional health economic evidence required? The CMC team heard positive news at the ESC
A sign that GLP-1 competition has entered the second half is that the narrative of clinical value has been fully established; the next phase will be a contest of formulation technology, production capacity, patient adherence, and differentiated endpoints. Now that semaglutide and tirzopotide have demonstrated cardiovascular benefits and weight loss effects, it will be difficult for later entrants to differentiate themselves in the market if they can only replicate the same clinical endpoints. The real competition is shifting to manufacturing and delivery.
In terms of formulation technology, longer-acting formulations (such as those administered monthly or at even longer intervals) present new challenges for scale-up. Sustained-release microspheres, in-situ gels, or implantable formulations all require solutions for release curve uniformity, the impact of sterilization processes on polymers and proteins, and batch-to-batch consistency in large-scale production.Oral SNACs also face the issue of batch-to-batch variability in bioavailability. Absorption-enhancing technologies for GLP-1 formulations (such as oral semaglutide) (
production capacity is a more pressing challenge. It will take 2 to 3 years. During this period, CDMOs and supply chain companies that can provide competitive high-capacity peptide solutions will gain significant market opportunities.The annual production capacity demand for GLP-1 APIs has expanded from the metric-ton range to the hundred-metric-ton range. Scaling up solid-phase peptide synthesis involves optimizing multiple aspects, including resin loading capacity, coupling efficiency, and purification yield. Both Novo Nordisk and Eli Lilly are investing heavily in capacity expansion, but ramping up production typically requires
| Competitive Dimensions | First Half (What Has Happened) | Second Half (Currently Unfolding) | Implications for the CMC Team |
| Clinical Endpoints | Reduction in MACE; Weight loss | Subgroup analysis; long-term safety | Clinical data required to support indication expansion |
| Dosage Form | Weekly injection pen | Monthly formulation/oral/implant | Development of new dosage form processes |
| Production Capacity | Tonne-scale API | 100-metric-ton-scale API | Scale-up; Supply Chain Resilience |
| Adherence | Convenience of injection pens | Digital management; side effect control | Combination drug-device products |
| Differentiation | Target selection (GLP-1/GIP/GLP-1+GIP) | Combination Formulations; Patient Stratification | Quality Control of Multicomponent Formulations |
Compound or multi-target designs significantly increase the complexity of quality control. Combinations of amylin analogs and GCG agonists in a single formulation require separate characterization and joint evaluation of each component’s release profile, stability, and interactions. This poses entirely new requirements for analytical methods and release criteria. Competition over differentiated endpoints is also accelerating. While GLP-1 combined with other mechanisms (such as telapreotide, which is already a dual-target molecule) may
Competition based on differentiated endpoints is also accelerating. Now that MACE reduction has become the standard for GLP-1, new competitors need to demonstrate differentiated benefits in specific subgroups—such as in HFpEF patients, where CMC teams provide supporting analytical method development.Improving cardiac remodeling parameters in patients with HFpEF, slowing renal function decline in patients with CKD, or improving liver fibrosis in metabolic and alcoholic steatohepatitis (MASH). The clinical design, biomarker selection, and regulatory communication for these differentiated endpoints all require
Do longer-acting formulations present new scaling challenges? The answer is yes.The shift from weekly to monthly formulations means that the dose per injection needs to increase by about fourfold, which places new demands on the capacity of injection pens, the acceptability of the injection volume, as well as the concentration and stability of the formulation. High-concentration protein formulations are prone to aggregation and viscosity issues, which affect the ease of injection and the patient experience. In combination formulations, the coexistence of multiple components may lead to interactions, affecting their respective release profiles and stability.
CDMOs and supply chain companies play a critical role in this competition. When a pharmaceutical company’s own production capacity is insufficient to meet market demand, the production capacity flexibility of CDMOs serves as a crucial buffer. However, the qualification thresholds for large-scale peptide CDMOs are extremely high—requiring GMP-grade peptide synthesis facilities, high-purity purification capabilities, and a complete capability chain for formulation filling and packaging.Globally, there are no more than 20 CDMOs capable of supplying GMP-compliant peptide APIs at the kilogram- to metric-ton scale, and production capacity typically must be booked 12 to 18 months in advance.
Do longer-acting formulations present new scale-up challenges? Do combination or multi-target designs increase the complexity of quality control? The answers to these two questions directly impact the technical barriers to entry in the second half of the GLP-1 race.Injectable formulations must address viscosity and aggregation issues in high-concentration protein formulations; oral formulations must address batch-to-batch variability in bioavailability; and combination formulations must address release profile matching and interactions among multiple components. Behind every formulation technology challenge lies a comprehensive set of analytical method development and stability study protocols.
The expansion of production capacity also places new demands on supply chain resilience. The global supply of peptide APIs is concentrated in the hands of a few CDMOs; a production halt or quality issue at any single supplier could lead to a global disruption in GLP-1 supply. Pharmaceutical companies need to establish dual- or multi-source supply strategies and maintain safety stock for critical raw materials. The assessment of supply chain resilience should be incorporated into the overall risk management framework of GLP-1 projects.
3.2 As AOCs and small nucleic acids enter the cardiovascular field, analytical characterization will become part of the platform’s value
For an AOC platform to successfully navigate the path from concept to product in cardiovascular indications, the establishment and refinement of analytical characterization methods are an unavoidable step. For industry experts, understanding the actual technical barriers behind AOC is a better indicator of the platform’s long-term value than knowing the transaction amount.
| Quality Attribute Categories | Specific Indicators | Current Analytical Methods | Technical Challenges | Impact on Platform Value |
| Sequence-Related Impurities | Shortmers, Longmers, and Sequence Deletions | Ion-exchange chromatography (IEX), LC-MS | Limited MS resolution for long-chain oligonucleotides affects quantitative accuracy | Directly affects product purity and batch consistency; these are core indicators for regulatory release |
| Coupling quality | Drug-to-antibody ratio (DAR), conjugation site distribution, free oligonucleotide proportion | Hydrophobic Interaction Chromatography (HIC), LC-MS Peptide Map | Conjugation site polymorphism leads to complex peak shapes and makes quantification difficult | The DAR distribution directly impacts efficacy and safety and is the core quality attribute that distinguishes AOCs from conventional antibodies |
| Antibody-Related Impurities | Aggregates, fragments, host cell proteins, and detached Protein A | SEC-HPLC, CE-SDS, ELISA | The physicochemical properties of antibodies change after conjugation, so existing methods may not be applicable | The risk of immunogenicity is directly related to impurity levels |
| Tissue distribution | Drug concentration in target tissues (muscle/myocardium) and off-target tissue distribution | Radioactive labeling + tissue autoradiography, qPCR | Ethical and practical limitations of myocardial sampling; sensitivity of the limit of quantification | Tissue distribution data are the key evidence demonstrating delivery efficiency and directly support clinical translation |
| Stability | Freeze-thaw stability, long-term storage stability, and rate of conjugate bond cleavage | Accelerated stability testing + real-time stability monitoring | There is a lack of predictive models for the kinetics of conjugate bond cleavage under different conditions | This affects shelf-life determination and storage/transport conditions, and is critical to commercial viability |
Each parameter in this table corresponds to a regulatory question that must be addressed as an AOC progresses from preclinical development to market launch. Taking conjugation quality as an example, the distribution of the drug-to-antibody ratio (DAR) directly affects efficacy and safety—molecules with excessively high DAR may exhibit increased toxicity due to an excess of oligonucleotides, while those with excessively low DAR may demonstrate suboptimal efficacy due to insufficient payload.Similar to ADCs (antibody-drug conjugates), the DAR distribution of AOCs must be precisely characterized using analytical methods, and acceptable ranges must be established in release criteria. However, unlike ADCs, the physicochemical properties of oligonucleotides as payloads differ significantly from those of small-molecule toxins; existing ADC analytical methods cannot be directly applied and require the development of tailored methods.
The control of sequence-related impurities presents another technical hurdle. During the solid-phase synthesis of oligonucleotides, the efficiency of each coupling reaction cannot reach 100%; unreacted sequences form shortmers (short chains missing one or more nucleotides), while overreaction results in longmers (long chains with one or more extra nucleotides).These impurities are structurally highly similar to the target sequence, making them difficult to separate and purify. For AOC, if these oligonucleotide impurities are not sufficiently removed prior to coupling, they will be carried over into the final product, affecting its purity and safety.
Obtaining tissue distribution data poses a unique challenge for the AOC platform in cardiovascular indications. In DMD trials, muscle biopsies can provide direct evidence of local drug concentrations. However, in cardiovascular indications, the ethical and practical limitations of myocardial biopsies are far greater than those for skeletal muscle.This means that when transitioning to cardiovascular applications, the AOC platform must develop alternative methods for assessing tissue distribution—such as indirectly evaluating myocardial delivery efficiency through imaging techniques (e.g., PET probes) or inferring target tissue effects based on changes in circulating biomarkers. The establishment and validation of these methodologies are integral to the platform’s value.
From a regulatory communication perspective, this means that AOC developers need to engage in more pre-IND and Type B meetings with regulatory authorities to reach consensus on analytical method validation, release criteria, and stability study protocols. Such communication requires robust data support, and the generation of this data, in turn, depends on the maturity of analytical methods—creating a cycle of mutual dependence. As a novel class of conjugates, AOCs currently lack mature guidance documents.The EMA’s 2024 draft guidance on oligonucleotides and existing guidance on antibody drugs cover two components of AOCs, respectively, but the regulatory framework for the conjugate as a whole is still evolving.
As AOCs and small nucleic acids enter the cardiovascular field, analytical characterization will become an integral part of the platform’s value. The reason is simple: when the tissue selectivity, conjugation consistency, and long-term safety of a delivery platform have not yet been fully validated in clinical trials, the depth and precision of analytical data serve as the core basis for regulatory authorities and collaborators to assess the platform’s maturity.
Sequence-related impurities are a category of impurities specific to oligonucleotides. In solid-phase synthesis, the efficiency of each coupling reaction cannot reach 100%; unreacted sequences exist as short chains such as n-1 and n-2 (shortmers), while over-coupling or side reactions produce long chains such as n+1 and n+2 (longmers).In addition, incomplete base modification, incomplete deprotection, and incomplete oxidation of the phosphodiester bond can all lead to a series of structure-related impurities.For naked oligonucleotide drugs, well-established methods (such as ion-exchange HPLC and capillary electrophoresis) are available for the separation and quantification of these impurities. However, in AOC, the impurity profiles of free oligonucleotides and conjugated oligonucleotides must be characterized separately.
| Dimensions of Analytical Characterization | Key Metrics | Common Methods | Challenges Specific to AOC |
| Conjugation Consistency | DAR Distribution; Coupling Sites | HIC-HPLC; Mass Spectrometry | Large molecular weight of oligonucleotides; insufficient resolution |
| Free oligonucleotides | Percentage of unconjugated oligonucleotides | SEC-HPLC; AEX-HPLC | Separation from conjugated products |
| Oligonucleotide impurities | Shortmers/Longmers | IP-RP-HPLC; CGE | Separate quantification of free vs. conjugated impurities |
| Antibody-related impurities | Aggregates; Fragments | SEC-HPLC; CE-SDS | Conjugation may alter antibody conformation |
| Tissue distribution | Concentration in target tissues; off-target distribution | Radiolabeling; qPCR | Methods for Translating from Preclinical to Clinical Applications |
Controlling the coupling ratio (DAR distribution) begins with the selection of the coupling chemistry for AOCs, which requires striking a balance between process controllability, production costs, and product performance. Common coupling strategies include cysteine coupling (via reduction and subsequent formation of interchain disulfide bonds), lysine coupling (via random binding to surface lysine residues), and site-specific coupling(via the engineered introduction of specific or non-natural amino acids). Each strategy exhibits distinct DAR distribution characteristics: cysteine coupling typically yields a broad DAR distribution (0–8); lysine coupling results in an even broader distribution that is more difficult to control; and site-specific coupling can achieve a narrow DAR distribution but requires additional engineering steps.
Control of free oligonucleotides is another key release criterion. Unconjugated free oligonucleotides not only reduce the payload ratio but can also enter cells via off-target pathways, increasing the risk of off-target silencing.Release criteria must establish an upper limit for free oligonucleotides, which requires a purification process capable of effectively removing free components; however, excessive purification steps may reduce yield and introduce new process variations.
The stability monitoring system also needs to be redefined. Changes that may occur in AOCs during storage include: antibody structural degradation (deamination, oxidation, aggregation); oligonucleotide degradation (hydrolysis of phosphodiester bonds, depurination); cleavage of the conjugation bond (leading to an increase in free oligonucleotides); and conformational changes affecting binding activity.Stability monitoring methods must be capable of simultaneously tracking these degradation pathways and establishing a correlation with clinical efficacy.
To demonstrate to a specialized audience that the authors understand the true challenges behind platform-based manufacturing, it is necessary to delve into specific technical metrics. For example, quantitative analysis of sequence-related impurities requires the use of ion-pair reversed-phase HPLC (IP-RP-HPLC) or capillary gel electrophoresis (CGE). These methods have been thoroughly validated for the analysis of naked oligonucleotides.However, in AOC, impurity analysis of free oligonucleotides can be performed using these methods after removing the conjugated components, whereas impurity analysis of conjugated oligonucleotides requires the development of new methods. This is because the molecular weight and charge characteristics of the conjugated products differ from those of naked oligonucleotides, and existing chromatographic conditions cannot effectively separate them.
Obtaining tissue distribution data is another technical challenge in AOC platform validation. Preclinical studies typically use radiolabeled oligonucleotides (such as 3H- or 14C-labeled oligonucleotides) to track drug distribution in vivo. However, radiolabeling can alter the physicochemical properties of oligonucleotides, thereby affecting delivery efficiency.Alternative methods, such as qPCR or hybridization ELISA, can detect oligonucleotide concentrations in tissues, but their sensitivity and specificity are insufficient to distinguish between the intact drug and its degradation products. The translation of tissue distribution validation methods from the preclinical to the clinical setting is an analytical methodological challenge that the AOC platform must address.
Translating the technology into specific metrics: sequence-related impurities, shortmers, longmers, conjugation ratio, free oligonucleotides, antibody-related impurities, tissue distribution, and stability. These metrics constitute the core data set for AOC analytical characterization. Each metric requires a validated analytical method, clear release criteria, and a stability monitoring protocol.The article does not need to read like a technical textbook, but it should convey to professional readers that the author understands the real challenges underlying platform operations.
Acquiring and interpreting tissue distribution data: qPCR methods can detect oligonucleotide concentrations in tissues, but extraction methods must be optimized to remove conjugated components that may interfere with PCR. Hybridization ELISA can provide higher specificity, but its sensitivity is insufficient to detect low-abundance target tissue distribution. Each method has its advantages and limitations; the AOC platform must establish multiple complementary analytical methods to comprehensively evaluate tissue distribution.Key Steps in AOC Platform Validation. In preclinical studies, radiolabeling methods can provide quantitative data on systemic distribution but cannot distinguish between the intact drug and its degradation products.
3.3 There Is Still Room for Small-Molecule and Large-Molecule Cardiovascular Drugs, but They Cannot Survive on Old Narratives Alone
The key lies in traditional drugs needing to demonstrate something more substantial than merely a “well-established mechanism of action”: clearer patient stratification, more explicit value in combination therapy, or superior safety data.The brighter the spotlight on GLP-1 and nucleic acid drugs, the more the narrative space for traditional small-molecule and large-molecule cardiovascular drugs is squeezed. However, this does not mean that traditional drug forms have lost their value—on the contrary, small molecules and antibody drugs still hold an irreplaceable position in targets and indications not covered by GLP-1.
| Drug Formulations | Areas Under Pressure from GLP-1/AOCs | Areas Where Irreplaceable Value Remains | Dimensions of Narrative That Need to Be Supplemented |
| Small-Molecule Cardiovascular Drugs | Lipid-lowering (the PCSK9 segment is covered by siRNA), antiplatelet (some indications are being replaced by cardiovascular benefits of GLP-1) | Anticoagulation (DOACs still have essential demand), Arrhythmias (ion channel modulation), Acute-Phase Treatment (thrombolysis, vasodilation) | Safety data for combination use with GLP-1, patient stratification based on specific genotypes/phenotypes, and differentiated positioning between acute and chronic indications |
| Large-Molecule Cardiovascular Drugs | PCSK9 antibodies face competition from siRNAs such as Inclisiran in terms of dosing frequency | Complement pathway (cardiovascular inflammation), coagulation factors (cardiovascular complications associated with hemophilia), rare inherited cardiovascular diseases | Evidence of efficacy advantages beyond dosing convenience; demonstration of the undruggability of the target |
| Gene therapy | No direct competition yet (in earlier stages of development) | Hereditary cardiovascular diseases (e.g., hypertrophic cardiomyopathy with MYBPC3 mutations) | Long-term safety follow-up data, feasibility of administration methods |
| Cell Therapy | No direct competition yet | Myocardial regeneration (iPSC-derived cardiomyocytes), immunomodulation (cardiovascular inflammation) | Control of Oncogenic Risk, Survival Rate, and Functional Integration of Transplanted Cells |
Taking the PCSK9 field as an example, as PCSK9 monoclonal antibodies, evolocumab and alirocumab have already demonstrated superior lipid-lowering efficacy compared to Inclisiran (rather than going head-to-head with GLP-1 or siRNA on the same dimension, they identify scenarios not covered by those therapies and establish a differentiated positioning based on patient stratification and combination therapy data).siRNA) in terms of dosing frequency, shifting from biweekly subcutaneous injections to once every six months. However, monoclonal antibodies still hold their own ground: in the context of early intensive lipid-lowering therapy following acute coronary syndrome (ACS), they retain advantages in terms of onset of action and dose flexibility. Traditional drugs should not aim to compete head-to-head with
anticoagulants (DOACs) is another area where traditional small molecules still have a strong unmet need. Direct oral anticoagulants (DOACs) such as rivaroxaban, apixaban, and dabigatran remain the standard of care for the prevention and treatment of atrial fibrillation, deep vein thrombosis, and pulmonary embolism; the cardiovascular benefits of GLP-1 agonists cannot replace the need for anticoagulation.However, DOACs face challenges in managing bleeding risks and adjusting dosages for specific populations (such as the elderly and those with renal impairment)—issues that require real-world data and patient stratification to address.
For developers of traditional cardiovascular drugs, GLP-1 data present an opportunity to “differentiate” their own products.Every time you hear about positive results for a GLP-1 or nucleic acid drug, you should ask: Does this result squeeze the space for my pipeline? If so, in which patient subgroups or combination therapy scenarios does my drug still hold irreplaceable value? If not, are there opportunities to explore combination therapies? This kind of cross-disciplinary thinking has greater long-term value than simply tracking trends in a specific field. The strategy for attending the ESC Congress 2026 should be “listen
Action Recommendation: Before attending, traditional cardiovascular drug teams should list the three indication areas in their pipeline most at risk of being squeezed by GLP-1 or nucleic acid drugs, and prepare two response strategies for each area: “combination therapy opportunities” and “differentiated patient stratification.” During the conference, focus on subgroup analyses of GLP-1 in these indications to identify evidence-based anchors demonstrating that their own drugs still hold irreplaceable value.
There is still room for small-molecule and large-molecule cardiovascular drugs, but they cannot survive by relying solely on old narratives. The spotlight on GLP-1 and nucleic acid therapeutics will squeeze the narrative space for conventional projects; traditional cardiovascular drugs must demonstrate clearer patient stratification, value in combination therapy, or safety advantages to maintain their market position.
Among small-molecule cardiovascular drugs, the development of oral PCSK9 inhibitors is progressing. If oral PCSK9 inhibitors can achieve LDL-C-lowering effects comparable to those of injectable formulations, they will significantly alter the prescribing pathway for lipid-lowering therapy, shifting from specialist-administered injections to general practitioner-prescribed oral medications. However, this requires resolving formulation challenges such as oral bioavailability, first-pass hepatic metabolism, and drug interactions.
Among macromolecular cardiovascular drugs, there remains room for expanding the indications of antibody therapies. The preventive value of anti-RSV antibodies in patients with heart failure complicated by respiratory infections, the application of anti-IL-6 antibodies in managing inflammation following myocardial infarction, and the efficacy of anti-ANGPTL3 antibodies in severe hypertriglyceridemia are all areas worthy of attention.However, when assessing the commercial value of these projects, the competitive impact of GLP-1 in the same patient population must be taken into account. If GLP-1 can simultaneously improve multiple cardiovascular and metabolic risk factors, it will become more difficult to differentiate single-target antibody drugs.
| Drug Class | Advantages | Facing Competition from GLP-1/AOC | Differentiation Strategies |
| Small-molecule lipid-lowering drugs | Convenient oral administration; low cost | GLP-1 Improves Lipid Profile | Oral PCSK9 inhibitors; combination therapy |
| Small-molecule anticoagulants | Oral administration; well-established clinical experience | Indirect competition | Safety in special populations; bleeding risk |
| Macromolecular antibodies | High specificity; long half-life | AOCs may target the same receptor | Combination therapy; use during the acute phase |
| RNAi drugs (liver-targeted) | Long-acting; products already on the market | Complementary to AOCs rather than in direct competition | Liver-targeted metabolic diseases |
Traditional cardiovascular drugs still hold considerable commercial value if they can find a niche in combination therapy. For example, does the combination of statins and GLP-1 agonists produce a synergistic effect? At the ESC Congress 2026, attention should be paid to whether new data on combination therapy involving PCSK9 inhibitors will be released. How should sequential treatment regimens for inclisiran be designed? Clinical validation of these combination strategies requires specifically designed studies, rather than simple post-hoc analyses. Attendees at
From a pharmacoeconomic perspective, traditional small-molecule drugs—such as ACEIs/ARBs and beta-blockers—offer cost advantages. This means the market for traditional cardiovascular drugs will not disappear, but their role in treatment pathways needs to be redefined.This will become even more pronounced against the backdrop of limited GLP-1 production capacity. When GLP-1 supply falls short of demand, payers will prioritize allocating GLP-1 to the highest-risk patients, while low- and medium-risk patients will still need to rely on statins,
The spotlight on GLP-1 and nucleic acid drugs will squeeze the narrative space for conventional therapies, but they will not completely replace traditional drugs. There are three reasons for this: First, GLP-1 production capacity constraints mean that not all potential patients can be covered in the foreseeable future, so low- and medium-risk patients will still need to rely on traditional drugs.Second, the cost structures of AOCs and small nucleic acids dictate that, in the short term, they can only be used for rare diseases or high-value indications; small molecules will continue to dominate the treatment of common cardiovascular diseases on a large scale. Third, combination therapy strategies require traditional drugs as the foundation of treatment regimens; GLP-1 and AOCs serve more as add-ons than as substitutes.
HoFH) or patients with specific genotypes (such as those with LDLR mutations). Regarding the value of combination therapy, the prominence of statins paired with PCSK9 inhibitors and GLP-1 with nucleic acid drugs will limit the narrative space for general programs; traditional medications must demonstrate clearer patient stratification, the value of combination therapy, or safety advantages. In terms of patient stratification, traditional lipid-lowering drugs can focus on patients with severe hyperlipidemia who are not adequately covered by GLP-1(for example, the triple-therapy regimen with inclisiran has become the standard of care for patients at extremely high cardiovascular risk). In terms of safety advantages, the clinical experience with traditional oral anticoagulants in specific populations (such as the elderly and those with renal impairment) remains irreplaceable by GLP-1 agonists.

4. bio meeting 2026: ESC Congress Attendance Strategy for Different Priorities for BD, R&D, and Investment Teams
At a conference with over 33,000 attendees, approaching it with a one-size-fits-all strategy is a waste of time for most people. For clinicians, the core issue is not “who should attend,” but rather what questions each category of attendee should bring to the conference and how to translate what they hear into actionable decisions afterward. The focus of CMC leaders rarely overlaps with that of investors. The needs of the BD team are entirely different.
The following breakdown of strategies for these three types of attendees should provide specific conference objectives and post-conference action frameworks tailored to each group’s decision-making context, rather than simply addressing “who should attend.”
Different attendees come to the ESC Congress 2026 with different objectives and should not navigate the event using the same approach.Clinicians need to select sessions from the perspective of translating evidence into prescribing behavior; pharmaceutical R&D and CMC teams need to plan their schedules from the perspective of translating data into process development; and BD professionals and investors need to filter information from the perspective of translating data into asset valuation. The focus areas, information processing methods, and post-conference actions of these three types of attendees are entirely different; using the same set of strategies will result in inefficient information gathering.
4.1 Clinicians and Medical Affairs Teams: Assess Whether Evidence Can Change Actual Prescribing Behavior
The core objective for clinicians and medical affairs teams attending the ESC is not to learn “which new drugs are in development,” but to determine “whether existing prescribing practices need to be adjusted.” The distinction between the two lies in the fact that the former involves information gathering, while the latter involves decision support. A valuable cardiovascular conference should enable attendees to leave with a new perspective on at least one clinical decision.
| Dimensions of Prescribing Decisions | Current Standard Regimen | Potential Directions for GLP-1 | Key Questions Attendees Should Ask |
| Prevention of High-Risk Cardiovascular Events | Statins + Antiplatelet Therapy + ACEI/ARB | Will GLP-1 Be Included in Primary/Secondary Prevention Regimens? | Do the SELECT subgroup data cover different types of cardiovascular events? What are the expectations for guideline updates? |
| Heart Failure with Preserved Ejection Fraction (HFpEF) | Diuretics + Management of Comorbidities (No Drugs Available to Improve Prognosis) | Will GLP-1 become the first drug to improve prognosis in HFpEF? | Do the STEP-HFpEF extension data provide evidence for hard endpoints? What is the degree of symptom improvement? |
| Obesity-Related Cardiovascular Risk | Lifestyle interventions ± weight-loss medications (limited efficacy) | Will GLP-1 elevate obesity from a “risk factor” to a “treatable target”? | Is there a dose-response relationship between weight loss and the reduction in cardiovascular events? What is the risk after discontinuation? |
| Diabetes with concomitant cardiovascular disease | SGLT2 Inhibitors (preferably those with evidence of cardiovascular benefit) | GLP-1 vs. SGLT2: Which is the first-line choice? Combination therapy? | Safety and efficacy of combining these two classes of drugs? Selection strategies across different stages of CKD? |
| Cardiovascular protection in chronic kidney disease | ACEI/ARB + SGLT2 Inhibitor | Should GLP-1 (FLOW trial-positive) be included in cardiovascular protection regimens for CKD? | GLP-1 Dose Adjustment and Safety Across Different eGFR Tiers? |
Each row in this table corresponds to a real-world prescribing decision point. If GLP-1 can demonstrate data on harder endpoints in the HFpEF population, it may fill this therapeutic gap. However, clinicians need to ask: Taking HFpEF as an example, there are currently almost no drugs in clinical practice that improve prognosis in heart failure with preserved ejection fraction—diuretics can only alleviate symptoms, and evidence for SGLT2 inhibitors is still accumulating.Has the symptomatic improvement (KCCQ score) observed in STEP-HFpEF translated into benefits in hard endpoints (death and hospitalization for heart failure)? If not, is symptomatic improvement alone sufficient to change prescribing behavior?
The Medical Affairs team’s role is more focused on translation: turning the data presented at conferences into evidence packages that internal teams can use. This includes compiling the primary endpoints and subgroup analyses from key trials, updating data citations in medical materials, and preparing FAQs for the sales team to address customer inquiries.The most important task for the Medical Affairs team at the ESC is to identify those “gray areas” where the data is positive but the clinical translation is not yet clear. These areas are often also of interest to competitors’ medical teams; whoever is first to develop a clear scientific narrative will gain the upper hand in physician education.
For clinicians, it is also important to pay attention to a dimension that is often overlooked: the pace and direction of guideline updates. Discussions on ESC guideline updates typically begin within a few months of the Hot Lines session. If attendees can anticipate the potential direction of these changes in advance, they can prepare earlier in their clinical practice. Specifically, they should pay attention to the position statements and consensus documents released by the various ESC working groups during the conference, as these documents often serve as a prelude to guideline updates.
Action Recommendations: Before attending the conference, clinicians should list the five most common cardiovascular prescribing dilemmas they encounter in their outpatient practice and identify corresponding research presentations for each dilemma in the conference agenda. Within 48 hours after the conference, they should assess whether any of these five dilemmas require an adjustment to prescribing strategies based on new data. Medical affairs teams should complete an internal evidence update package within one week after the conference, including summaries of key data, subgroup analysis tables, and FAQ documents.
The core mission for clinicians and medical affairs teams attending the ESC Congress 2026 is to determine whether new evidence is sufficient to change actual prescribing behavior. This is a practical judgment, not an academic one—the fact that data is effective in a strictly controlled clinical trial setting does not mean it is equally applicable in community hospitals, primary care clinics, or long-term care facilities.
Changes in prescribing practices require three conditions to be met: the evidence must be sufficiently strong, the guideline recommendations must be sufficiently clear, and safety and adherence must be acceptable in the real world. While 20% of the evidence comes from hard endpoints, real-world evidence (RWE)—which is typically derived from electronic health record databases or prescription tracking systems—can more accurately reflect the performance of GLP-1 in real-world clinical settings.The SELECT study demonstrated a reduction in MACE. However, gastrointestinal adverse reactions associated with GLP-1, local reactions at the injection site, and logistical requirements for cold-chain storage may all reduce prescription rates and treatment adherence. Medical affairs teams should check whether real-world data is available on the ESC (
| Conditions for Prescribing Changes | Data Required | ESC 2026 Focus Areas | Post-Conference Actions |
| Strength of Evidence | Primary Endpoint: MACE; Subgroup Analysis | Hot Lines Announcements | Internal Medical Assessment |
| Guideline Recommendations | Draft ESC Guidelines Update | Late-Breaking Science | Preparing for Guideline Changes |
| Real-World Safety | ADR Reporting; Treatment Retention Rates | RWE Oral Presentations/Posters | Updating Safety Communication Materials |
| Adherence Data | 12-Month Treatment Retention Rate | Real-world cohort studies | Patient Support Program Design |
For heart failure specialists, data on HFpEF warrant particular attention. There are currently no effective medications to improve prognosis in HFpEF; if GLP-1 can demonstrate improvements in hard endpoints (cardiovascular death or hospitalization for heart failure) in the HFpEF subgroup, this would represent a major breakthrough in heart failure treatment. The STEP-HFpEF series of studies has already shown signs of symptom improvement, but there remains a significant gap between symptom improvement and improved prognosis.GLP-1 in heart failure with preserved ejection fraction (
The Medical Affairs team also needs to monitor data on the interactions between GLP-1 and other cardiovascular medications. GLP-1 may affect gastric emptying, thereby influencing the absorption of oral anticoagulants, antiplatelet agents, or diuretics. In heart failure patients, volume changes induced by GLP-1 may necessitate adjustments to diuretic dosages. While these drug interactions may be strictly controlled in clinical trials, physicians need the appropriate knowledge to manage them in real-world prescribing practices.If the ESC publishes relevant studies or reviews, the Medical Affairs team should obtain them immediately and incorporate them into internal training materials.
Is the data sufficient to alter the management pathway for high-risk patients? This is the most critical question clinicians need to address after attending the ESC meeting.Management pathways for high-risk patients typically involve the combined use of multiple medications—antiplatelet agents, lipid-lowering agents, antihypertensives, anticoagulants, and metabolic modulators. If there is a risk of MACE, does adding GLP-1 to the existing pathway pose a risk of drug interactions? Is it necessary to adjust the doses of other medications? These practical questions require answers from dedicated clinical studies, rather than simple subgroup analyses. The data on GLP-1 presented at the ESC demonstrate that it can reduce the risk of MACE in high-risk patients.
Is this evidence sufficient to change the management pathway for high-risk patients? Can it support expanding use from specialists to broader clinical practice? The former question concerns the strength of the evidence, while the latter concerns feasibility.Changes to management pathways for high-risk patients require an upgrade in the recommendation level within clinical guidelines, which typically necessitates pooled evidence from multiple confirmatory studies. Expanding use from specialists to general practitioners requires simplifying the prescribing process, establishing safety monitoring standards, and providing patient education materials. The pace of these translational efforts is often much slower than that of the clinical data itself.
4.2 Pharmaceutical R&D and CMC Teams Must Break Down Key Data into Next-Step Development Questions
Pharmaceutical R&D and CMC teams must consider: “If the data holds up, where will the subsequent challenges in formulation, manufacturing, analysis, stability, and supply chain arise?” This ability to translate clinical signals into development challenges is the most important takeaway for R&D teams at the conference. The way CMC teams participate in the ESC should differ fundamentally from that of clinicians. Clinicians are concerned with “whether the data will change prescribing practices,” while R&D and CMC teams are concerned with
| after hearing positive clinical signals | Development Questions R&D Teams Should Ask | Manufacturing Questions the CMC Team Should Ask | Capacity issues the supply chain team should address |
| Positive results for new GLP-1 indications (e.g., HFpEF/CKD) | Are different dosage strengths required? Will the dosing regimen change? Are new PK/PD models needed? | Does the formulation for the new dosage form require adjustment? Does the stability protocol need to be redesigned? | Estimated increase in patient population due to the new indication? Can existing production capacity accommodate this? |
| Tirpepetide SURPASS-CVOT Non-Inferiority | How should the differentiated narrative for the dual-target approach be restructured? Are there pipeline adjustments for new target combinations? | Is it necessary to expand peptide synthesis capacity? Can the purification process support higher purity requirements? | Is the supply of dual-target APIs diversified? |
| Early signals for myocardial delivery of AOC | How should the target validation strategy be adjusted? Is it necessary to initiate pre-IND discussions for cardiovascular indications? | Does the conjugation process require cardio-specific optimization? Do the analytical methods cover distribution in myocardial tissue? | Is AOC’s CDMO capacity sufficient? Are there alternative suppliers? |
| New Data on Oral GLP-1 Formulations | Has oral bioavailability improved? What are the directions for optimizing the formulation of absorption enhancers? | What are the challenges in process scale-up for oral solid dosage forms? How should suppliers of SNAC-type absorption enhancers be managed? | What is the production capacity allocation strategy between oral and injectable formulations? |
| Early clinical signals for small nucleic acids in cardiovascular indications | Directions for siRNA sequence optimization? Clinical validation protocols for dosing frequency? | Is it necessary to scale up solid-phase synthesis? Do impurity profiling methods meet regulatory requirements? | What is the global supply landscape for oligonucleotide APIs? Are there regional supply risks? |
The value of this table lies in its breakdown of a “positive outcome”—using the example of a new GLP-1 indication—into specific issues across three dimensions: R&D, CMC, and supply chain.Approval for the HFpEF indication does not simply mean adding a line to the existing Wegovy label—the new indication requires different dosage strengths (such as a lower starting dose to reduce gastrointestinal intolerance), specific stability data for heart failure patients (such as compatibility testing with commonly used heart failure medications), and updates to the administration device instructions (such as adjusting the injection volume for patients with fluid retention due to heart failure).Each of these issues requires the R&D and CMC teams to begin assessment as soon as clinical signals are received.
The most common mistake CMC teams make at the ESC is treating the conference purely as a “medical affair,” merely taking notes on the sidelines without forming their own judgments.In fact, the CMC team should attend the conference with its own list of questions—for example: Do the stability data for the current development project support the dosing regimen for the new indication? Is the coverage of analytical methods sufficient to support the new release criteria? If a new formulation of a competitor’s product is launched, what insights does it offer for the formulation design? These questions may not be answered directly during the conference, but the data and discussions presented can provide input for the CMC team’s subsequent evaluation.
For R&D teams, another often-overlooked value lies in dissecting competitor data. When hearing positive data from a competitor, one should not merely record that “Competitor XX showed positive results in Indication YY,” but rather ask: How does this result affect the positioning of our own pipeline? Is there room to explore combination therapy? Are there aspects of the competitor’s clinical design that we can learn from (such as endpoint selection, enrollment criteria, or follow-up duration)?This kind of systematic competitive analysis often holds greater strategic value than simply tracking data.
Action Recommendation: Immediately after R&D and Hot Lines research presentations, record the corresponding R&D, CMC, and supply chain follow-up questions in a table. Within two weeks of the meeting, organize a cross-functional meeting to translate these follow-up questions into specific development action items and budget requests. Before attending the meeting, the CMC team should prepare a “Clinical Signals to Development Issues” conversion table (similar to the structure of the table above) and fill it out in real time during the meeting. Each
Pharmaceutical companies’ approach to R&D and the ESC Congress 2026 should shift from merely reviewing data to breaking down problems. Whenever a major data point is announced, we must ask: If this clinical signal holds true, where will the pressure fall—on formulation, manufacturing processes, analysis, stability, batch consistency, or the supply chain? This mindset of translating data into action is the core value of CMC teams attending the conference.
Take GLP-1, for example, where prescription volumes have grown more than tenfold. If data on GLP-1’s renal protective effects in patients with chronic kidney disease (CKD) is presented at the ESC Congress, the CMC team must immediately assess: Does the impact of renal function staging on drug exposure require adjustments to dosage strengths? Is it necessary to develop formulations with lower dosages? If GLP-1 is expanded to a broader population for primary cardiovascular prevention, by how much will annual production capacity requirements increase? Can the existing supply chain support
| Types of ESC Data | Follow-up Questions from the CMC Team | Impact on Subsequent Development | Time Sensitivity |
| GLP-1 CKD Subgroup | Dose adjustment; new strengths; renal safety | Development of New Dosage Forms | High |
| GLP-1 HFpEF data | Combination formulations; Drug interactions | Feasibility assessment of combination formulations | In progress |
| AOC Clinical Data | Coupling Process; Analytical Methods; Stability | Platform Technology Assessment | Chinese |
| New Indications for Small Nucleic Acids | Delivery efficiency; Tissue distribution; Safety | Investment Decisions for Delivery Platforms | Medium to High |
For AOC and small nucleic acid platforms, key information the CMC team needs to seek at the ESC includes: Have any new clinical data on extrahepatic delivery been published? What are the quantitative data on delivery efficiency (target tissue concentration vs. plasma concentration)? Are there any delivery system-related events among the safety signals? This information will directly influence a company’s decision to invest in AOC platform technology or enter into relevant CDMO service agreements.
R&D teams also need to monitor regulatory trends regarding analytical methods. The EMA and FDA are currently updating their guidance documents on oligonucleotide-related impurities. If there are workshops or satellite sessions at the ESC attended by regulatory representatives, the CMC team should prioritize participation; these events often reveal regulatory expectations for the analytical characterization of new technology platforms, and gaining this insight early can help avoid additional testing requirements during subsequent regulatory submissions.
If a clinical signal is confirmed, where will the subsequent pressures lie—in formulation, process, analysis, stability, batch consistency, or the supply chain? This framework for follow-up questions applies to all new data presented at the ESC. Given the derivative requirements for GLP-1’s CMC and clinical development, an assessment should be initiated immediately after the conference.Take HFpEF data as an example: if the signal is confirmed, subsequent steps will include developing special dosage forms suitable for heart failure patients (who are sensitive to volume changes and require slower dose titration), evaluating drug interactions between GLP-1 and diuretics and ARNIs, and establishing heart failure-specific safety monitoring protocols. These
If the clinical signal is confirmed, where will the subsequent pressures lie in terms of dosage forms, manufacturing processes, analysis, stability, batch consistency, and the supply chain? The answer to this follow-up question determines the product’s timeline from clinical trials to commercialization—typically 2 to 3 years.Taking AOC as an example, if the clinical signal for cardiovascular indications is confirmed, the following steps will be required: developing formulations suitable for chronic administration (such as pre-filled pens), establishing a GMP-grade conjugation process, developing a comprehensive analytical characterization method package, conducting process validation and design space studies, and establishing a dual-source supply for oligonucleotide raw materials. The total time required for these tasks is typically
4.3 BD Teams and Investors: Distinguish Between “Validated Assets” and “Platforms Driven by Hype”
Rational attendees should bring a set of evaluation frameworks to translate conference hype into quantifiable criteria for judgment. The greatest risk facing BD teams and investors at the ESC is making decisions at the peak of market sentiment rather than missing out on hot trends. Top clinical conferences in the cardiovascular field naturally amplify optimism—a positive “Hot Line” announcement can cause a company’s stock price to rise by 10% in a single day, but such short-term volatility often does not reflect the asset’s long-term value.
| Evaluation Dimensions | Validated Assets (Worthy of a Premium) | Platforms Fueled by Sentiment (Require Caution) | How to Gather Information at the ESC |
| Data Maturity | Positive Phase 3 primary endpoint results, with complete safety data | Positive Phase 1 proof-of-concept or Phase 2 alternative endpoint results | Consider the study design phase and endpoint type in the “Hot Lines” section |
| Indications Scalability | Consistent subgroup data across multiple indications, supported by biological plausibility | Overall positive results for a single indication, but inconsistent subgroup results or insufficient sample size | Focus on consistency in subgroup analyses and data patterns across indications |
| Regulatory Pathway | Pre-IND/End-of-Phase 2 meeting minutes are available, and the direction of regulatory discussions is clear | No communication with regulatory authorities has taken place, or the outcome of such communication is unclear | Focus on regulatory workshops and regulatory update reports during the conference |
| Competition from Similar Products | First-in-class or best-in-class with clear data to support the claim | Me-too projects lacking evidence of differentiation | Track all reports on similar targets and compare efficacy and safety data |
| Manufacturing barriers | A mature CMC team and stable supply chain, or a partnership with a high-quality CDMO | High reliance on CMC outsourcing, single supplier, and lack of autonomy in analytical methods | Monitor CMC workshops and supply chain industry developments held in conjunction with conferences |
| Transaction Rationale | Valuation is based on clinical data and commercialization prospects | Valuation is based on platform concepts and transaction momentum, lacking cash flow support at the product level | Analyze valuation multiples and term structures of recent transactions |
The core logic of this table is that an asset’s long-term value depends on the maturity of its data and manufacturing capabilities, not on conference buzz. Take the AOC platform as an example: How comprehensive was Novartis’s $12 billion acquisition of the CMC team? Have analytical methods been established? If the answers to these questions are mostly “still in progress” or “not disclosed,” then the platform’s valuation relies more on expectations than on validation.The Avidity deal reflects the market’s high expectations for extrahepatic delivery platforms. But beyond DMD, is there clinical evidence for other indications? Have efficiency data for myocardial delivery been made public? When evaluating similar platforms, the BD team should ask: Aside from
The most common pitfall investors fall into at the ESC is equating positive results from Hot Lines—such as a reduction in MACE—with commercial success.A 20% result is academically significant, but the path from results to product launch involves multiple stages, including formulation optimization, long-term safety follow-up, regulatory communication, pricing negotiations, and health insurance access. Each stage could prevent clinical advantages from translating into commercial returns. Investors should evaluate ESC data within the context of the entire development chain, rather than focusing solely on whether a single endpoint is positive or negative.
Another aspect worthy of BD teams’ attention is the timing of the transaction. After positive data is released at an ESC, the valuation of related assets typically surges in the short term.If a BD transaction is initiated at the peak of market sentiment following the data release, the buyer often has to pay a higher premium. Conversely, if the team positions itself in advance—by making an informed prediction based on an understanding of the trial design prior to the data release, or by initiating negotiations during the cooling-off period after the data release (when valuations typically return to rational levels 2–3 months later)—the transaction can be completed at a more reasonable price. Seizing this timing window requires the BD team to conduct an in-depth, preemptive analysis of the ESC’s agenda and study design.
Action Recommendations: Before attending the meeting, BD and investment teams should compile a list of key target assets (3–5) to focus on and complete the evaluation across the six dimensions in the table above for each asset. Update the data during the meeting and finalize valuation adjustments and timing assessments within two weeks after the meeting. Avoid making any investment or transaction decisions within 48 hours of the Hot Lines release—the peak of market sentiment is not an optimal time for decision-making.
Both business development professionals and investors tend to assign valuation premiums to GLP-1—one of the most common mistakes made at AOC, the Small Nucleic Acids Conference, and ESC Congress 2026 is allowing conference hype to drive valuation judgments.Data released during “Hot Lines” sessions can cause a company’s stock price to fluctuate by more than 10% within 24 hours. However, attendees should focus on data maturity, indication scalability, regulatory pathways, competition from similar products, and manufacturing barriers—rather than transaction amounts or conference hype.
Assessing data maturity requires distinguishing between several levels: Phase I clinical data primarily address safety and tolerability; Phase II data begin to demonstrate efficacy signals but have limited sample sizes; Phase III data provide confirmatory evidence to support regulatory submissions. If an AOC project with only Phase I/II data is presented as a Hot Lines presentation at the ESC Congress, the BD team must carefully evaluate the reproducibility of its clinical signals—the probability that early-stage clinical efficacy signals will fail in Phase III trials remains high.
| Evaluation Dimensions | Signals Driven by Emotion | Validated Asset Signals | BD Action Recommendations |
| Data Maturity | Phase I Data Used as Proof of Concept | Phase III Hard-Endpoint Data | Distinguishing Between Proof of Concept and Confirmation |
| Indications Expansion | Claims to cover multiple indications | Clinical data spanning multiple indications | Evidence validating tissue selectivity |
| Regulatory Pathway | Claim of a Type B meeting having taken place | Pre-IND/End-of-Phase-2 communication letters available | Request to review meeting minutes |
| Competitors in the Same Class | Claim of “first-in-class” | Supported by patents and literature searches | FTO and competitive pipeline analysis |
| Manufacturing barriers | Claim of an exclusive process | Approved processes and release criteria | CMC audit |
Indication scalability is a key variable in platform valuation. While clinical data has been presented for DMD, its cross-indication value remains hypothetical.If the AOC platform is limited to BD, the team should ask: Does the platform have data on delivery efficiency across tissues? Is there proof of concept across targets? Is there a clear technical roadmap explaining the transition from muscle targeting to myocardial targeting? If the answers to these questions are vague, the platform’s valuation premium should be discounted.
Manufacturing barriers are the dividing line between platform value and product value.The availability of a GMP manufacturing facility. If an AOC platform possesses only unique antibody designs and oligonucleotide sequences but lacks established, mature conjugation processes, analytical methods, and scaled-up production capacity, its moat is shallow. CMC maturity should be evaluated as an independent dimension during due diligence—including the status of process validation, analytical method coverage, and the completeness of stability data packages. When evaluating platform deals, the BD team should factor in
2 to 3 years. GLP-1 candidates often command valuation premiums, but attendees should examine data maturity, indication scalability, regulatory pathways, competition within the class, and manufacturing barriers. Of these five dimensions, manufacturing barriers are the most easily overlooked.If an AOC platform possesses only unique antibody designs and oligonucleotide sequences but has not established GMP-grade conjugation processes and analytical methods, its commercialization timeline will be delayed by CMC issues. AOC, small nucleic acids, and CMC maturity should be treated as independent valuation discount factors. When evaluating platform deals, BD teams should consider
GLP-1 platforms often command valuation premiums, but attendees should focus on data maturity, indication scalability, regulatory pathways, competitive landscape, and manufacturing barriers—rather than solely on transaction amounts or conference buzz. Transaction amounts reflect supply-and-demand dynamics at a specific point in time and are not necessarily an accurate measure of a platform’s intrinsic value. Conference buzz reflects short-term sentiment and is not necessarily a reliable indicator of long-term trends.True valuation should be based on a systematic assessment of the five dimensions mentioned above, taking into account the interactions among them. AOC, small nucleic acids,

5. The Most Valuable Takeaway from the bio meeting 2026 Is Not Conclusions, but a Set of Judgment Questions
A good conference should leave attendees not with a few “conclusions,” but with a set of well-crafted judgment questions. Conclusions become outdated, but good questions can continue to guide decision-making.The questions in the following three areas—GLP-1, AOC and small nucleic acids, and cardiovascular drugs in general—each correspond to a set of decision-making frameworks that can be continuously applied in subsequent work. For attendees of the ESC Congress 2026, leaving Munich with a clear list of questions is far more meaningful than remembering a dozen or so positive results.
After reading this article, readers should know what questions to ask at the conference, rather than simply remembering where it is being held. The following three subsections provide a set of evaluative questions, not conclusions. The answers to these questions will need to be gradually gathered on-site at the ESC Congress 2026 by attending presentations, reviewing posters, and engaging with researchers.
5.1 Regarding GLP-1: How Much Additional Systemic Value Beyond Cardiovascular Benefits Can It Demonstrate?
The SELECT trial has established this. But the next question is more challenging: How far can this systemic value be extended? The following list of follow-up questions serves not as a one-time checklist, but as an evaluative framework that can be used to track progress over the next two to three years. The value of GLP-1 agonists in cardiovascular outcomes has already been demonstrated through
| Dimensions for Further Inquiry | Specific Questions | Current State of Evidence | Assessment Criteria |
| Endpoint Robustness | Are there new MACE hard endpoint data? Are there all-cause mortality data? | SELECT has demonstrated a 20% reduction in MACE; there is a trend toward reduced all-cause mortality, but it has not reached statistical significance | Positive hard endpoints (death/MI/stroke) > positive soft endpoints (symptoms/function) |
| Population Coverage | Does the efficacy data cover different BMI strata (>35 vs. <30)? Does it cover different diabetes statuses? | The SELECT trial enrolled participants without diabetes; the SUSTAIN-6 trial included a diabetes subgroup | Consistency across populations > Positive results in a single population |
| Heart Failure Subtypes | Is efficacy consistent between HFpEF and HFrEF? Are different formulations or doses required? | STEP-HFpEF focused on HFpEF; data on HFrEF are limited | Subtype-specific evidence supports separate indications |
| Long-term Safety | Are there data on pancreatitis, thyroid C-cell hyperplasia, and gastrointestinal events from follow-up periods of 5 years or longer? | SELECT had a median follow-up of 3.3 years; long-term data are still being collected | Acceptable long-term safety = supports long-term use for chronic conditions |
| Rebound after discontinuation | How do body weight and cardiovascular risk change after discontinuation? Is maintenance therapy necessary? | The STEP 1 extension phase showed a significant rebound in body weight after discontinuation | A clear maintenance therapy strategy = manageable adherence risks |
| Reimbursement Pressure | What is the pricing logic for GLP-1 in health insurance negotiations? Do cardiovascular indications change payers’ attitudes? | Wegovy is priced higher, and some health insurance plans restrict its prescribing scope | Approval of cardiovascular indications expands health insurance coverage = Raises the ceiling for commercialization |
| Combination Therapy | Safety of GLP-1 + SGLT2? Interactions between GLP-1 and oral anticoagulants? | Data on combination therapy is limited, but it is already being used in clinical practice | Sufficient safety data on combination therapy = expanded prescribing scope |
These seven dimensions form a comprehensive framework for evaluating the systemic value of GLP-1. Attendees at the ESC should seek out the latest GLP-1 cardiovascular data updates for each dimension and form their own conclusions after the conference. The three dimensions that warrant the most attention are: population coverage, long-term safety, and rebound effects upon discontinuation; together, these determine whether GLP-1 can evolve from a “weight-loss drug with cardiovascular benefits” into a “routinely prescribed cardiovascular preventive medication.”
If new data from the SELECT extension analysis emerges at ESC 2026—such as subgroup results stratified by BMI or updates on long-term safety—attendees should immediately update their assessments based on the table above.If the evidence for a particular dimension shifts from “limited” to “substantial,” the corresponding commercial expectations should be raised; conversely, if adverse signals emerge (such as reduced efficacy in specific subgroups or new safety signals), the market positioning of GLP-1 will need to be reassessed.
Regarding MACE, it is a composite endpoint comprising cardiovascular death, non-fatal myocardial infarction, and non-fatal stroke. However, different components may respond differently to GLP-1; if the reduction in MACE stems primarily from a decrease in strokes rather than a decrease in myocardial infarctions, its clinical significance and prescribing patterns will differ significantly.For GLP-1, the first question attendees should bring to the ESC Congress 2026 is: Is the endpoint sufficiently robust? Pay close attention to the specific data for each component of MACE when the Hot Lines results are released.
The second question is whether the efficacy extends across different BMI levels, diabetes status, and heart failure subtypes.Cardiovascular benefits are reduced in patients with a BMI below 30, which could impact obesity diagnostic criteria. If subgroup analyses from the SELECT study show that the reduction in MACE with GLP-1 is significantly smaller in the non-diabetic subgroup compared to the diabetic subgroup, the rationale for prescribing GLP-1 in individuals with metabolic syndrome but without diabetes will require more careful evaluation. Policy implications regarding GLP-1 prescribing eligibility. Similarly, if the non-diabetic subgroup’s
| GLP-1 Follow-Up Dimensions | Specific Issues | Current Evidence Status | ESC 2026 Expectations |
| Endpoint Rigidity | Is there a consistent reduction in all components of MACE across groups? | Overall MACE – 20% | Subgroup analysis data |
| Population Coverage | Non-diabetic; HFpEF; CKD subgroups | SELECT excluded certain populations | New Subgroup Analysis |
| Long-term Safety | Safety data for 5 years or more | Median follow-up of 3.3 years in SELECT | Extended follow-up cohort |
| Rebound After Discontinuation | Weight and CV Events After Discontinuation | Very limited data | Discontinuation follow-up study |
| Reimbursement rationale | Cost-effectiveness threshold | Significant Variations Across Countries | Health economic analysis |
The third point of inquiry concerns long-term safety and rebound effects following discontinuation. The median follow-up period in the SELECT study was approximately 3.3 years; for cardiovascular medications requiring lifelong use, this timeframe is insufficient to detect certain delayed adverse reactions, such as pancreatic cancer (which typically has an incubation period exceeding 5 years), thyroid C-cell hyperplasia (observed in animal studies but with limited human data), or the cumulative effects of prolonged use on bone density and muscle mass.If extended follow-up data from the SELECT study are presented at the ESC Congress, attendees should pay close attention to these late-onset safety signals.
Questions regarding reimbursement logic must also not be overlooked. If the Incremental Cost-Effectiveness Ratio (ICER) exceeds payers’ willingness-to-pay thresholds, prescription coverage will be limited even if the clinical evidence is robust. Attendees should pay attention to whether health economic analyses from different countries are presented at the ESC; these data will directly impact the accessibility of GLP-1 in various national markets. The annual treatment cost of GLP-1 remains significantly higher than that of traditional cardiovascular drugs in most countries. If health economic evaluations show that the cost-effectiveness ratio of GLP-1 (
Long-term safety, rebound effects after discontinuation, and reimbursement pressures may limit the availability of follow-up data beyond five years needed to confirm the risk of delayed adverse reactions.Weight regain following discontinuation requires dedicated post-discontinuation follow-up cohort studies to assess the rate of weight regain and the recurrence of cardiovascular events. Reimbursement pressures require health economic evaluations in each country to verify whether the ICER of GLP-1 falls within the payer’s willingness-to-pay threshold. Commercialization of GLP-1? These three factors constitute the final hurdle for GLP-1 to transition from clinical success to commercial success. Long-term safety requires
Are the endpoints sufficiently robust? Does efficacy extend across different BMIs, diabetic statuses, and heart failure subtypes? Will long-term safety, post-discontinuation rebound, and reimbursement pressures limit commercialization? These three questions form the comprehensive evaluation framework for GLP-1 at the ESC Congress 2026. The answers to each question require a comprehensive assessment of data from multiple studies, rather than relying on the overall results of a single study.
5.2 Regarding AOCs and Small Nucleic Acids: After Delivery Breakthroughs, Do Quality and Safety Hold Up Equally?
Breakthroughs in the extrahepatic delivery of AOCs and small nucleic acid drugs have opened up new possibilities, but the value of these platforms cannot be demonstrated by delivery alone. The following list of questions covers the key validation dimensions from platform to product.
| Follow-Up Dimensions | Specific Questions | Current Stage of Evidence | Evaluation Criteria |
| Cross-Indications Reusability | Does the AOC platform show clinical signals for indications other than DMD? Are there any animal models or early-stage clinical data for cardiovascular indications? | Positive DMD Phase 1/2 data; cardiovascular indications are still in the preclinical stage | At least one cardiovascular indication has entered the IND-enabling phase = preliminary validation of platform transferability |
| Tissue Selectivity | What is the delivery efficiency of AOC in cardiac muscle? How does it differ from skeletal muscle? What is the off-target tissue distribution? | Clinical data on skeletal muscle delivery are available; data on cardiac muscle primarily come from animal models | Myocardial delivery efficiency > 30% of that in skeletal muscle = warrants further cardiovascular development |
| Long-term administration risks | What are the immunogenicity data for repeated dosing? What is the incidence of anti-drug antibodies (ADAs)? What is the impact on delivery efficiency and safety? | ADA data are available from DMD trials, but long-term data are limited | Low ADA incidence with no impact on efficacy = Repeated dosing is feasible |
| Analytical Methods | Has a comprehensive analytical characterization method been established for the AOC? Have the analytical methods for conjugate quality, impurity profiles, and tissue distribution been validated? | Some methods draw on experience with ADCs and oligonucleotides; AOC-specific methods are still under development | Key analytical methods have been validated = supporting regulatory submissions and release |
| Release Criteria | Has consensus been reached with regulatory authorities regarding release criteria? What is the acceptable range for DAR? What are the impurity limits? | There are no AOC-specific guidelines yet; guidelines for oligonucleotides and antibodies are being referenced | Clear minutes from pre-IND meetings with the FDA/EMA = a clear regulatory pathway |
| Safety Evidence | Could myocardial delivery potentially trigger arrhythmias? Is there a risk of compensatory mechanisms following siRNA-mediated silencing of the target gene? | Preclinical safety data are limited; cardiotoxicity for specific targets still needs to be evaluated | Cardiovascular safety pharmacology studies passed = Safety foundation for advancing to clinical trials |
| Manufacturing Feasibility | Does AOC have GMP-grade manufacturing capacity? Can the batch size support Phase 3 and commercialization? | Avidity already has GMP manufacturing capacity, but the scale is limited | Successful GMP batch at Phase 3 scale = validation of commercial manufacturing feasibility |
These seven dimensions of inquiry form the three most critical ones—cross-indication reusability, tissue selectivity, and long-term administration risks—which determine whether AOC can establish a viable development pathway in cardiovascular indications. If the evidence for these three dimensions remains at the “preclinical” or “limited” stage, then the application of AOC in the cardiovascular field will remain at the conceptual stage, still a considerable distance from clinical translation.The complete validation pathway for the AOC platform, from “market buzz” to “product commercialization.”
Attendees at the ESC should focus on two key indicators: first, whether cardiovascular-related animal model data for AOCs or small nucleic acids are presented in satellite sessions or workshops; second, whether discussions regarding the direction of guidelines for oligonucleotide conjugates take place in regulatory workshops. Although these indicators do not constitute “Hot Lines”-level announcements, they provide direct reference value for assessing the expected timeline for the AOC platform in the cardiovascular field.
Regarding AOCs and small nucleic acids, the most critical question following delivery breakthroughs is whether quality and safety are equally well-established. If the AOC platform achieves targeted delivery to cardiac or skeletal muscle, this is only the first step. Attendees need to ask: What are the quantitative data on delivery efficiency? What is the ratio of drug concentration in the target tissue to plasma concentration? Is the distribution in off-target tissues acceptable?
Evidence of tissue selectivity is the cornerstone of the platform’s value. If the ratio is 1:1, it indicates that delivery remains non-specific, and the value of antibody-targeted delivery is limited. These quantitative data are typically not presented in detail during oral presentations at the conference; attendees should seek them out during the poster session or in post-conference discussions with researchers.If the AOC concentration in cardiac muscle is 10 times that in skeletal muscle and 5 times that in the liver, this indicates that the antibody-targeted strategy is effective. However, if the concentration ratio between cardiac and skeletal muscle is close to
| AOC Analysis Dimensions | Specific Questions | Evaluation Criteria | Risk Signals |
| Tissue Selectivity | Concentration Ratio of Target Tissue vs. Non-Target Tissue | >5:1 indicates effective targeting | <2:1 indicates nonspecific distribution |
| Long-term Administration Safety | ADA incidence; cumulative toxicity | ADA < 10%; no cumulative toxicity | ADA > 30%; cumulative liver and kidney toxicity |
| Maturity of analytical methods | Release criteria; stability indicator method | Complete analytical package available | Only partial methods available |
| Regulatory Communication | Pre-IND/Type B Feedback | Clear regulatory pathway | Uncertain regulatory feedback |
| Reusability | Cross-target; cross-indication evidence | Data for 2 or more targets | Validated for only 1 target |
Risks of Long-Term Administration All components of the AOC—including the antibody, oligonucleotide, and linker—may become immunogenic. Attendees should pay close attention to ADA data from early-phase clinical trials; if the ADA positivity rate exceeds 10%, the platform’s suitability for repeated dosing must be reevaluated. This assessment should consider both immunogenicity and cumulative toxicity.The incidence of ADA (anti-drug antibodies) may increase after repeated dosing, which not only affects efficacy but may also lead to immune complex-mediated tissue damage.
The maturity of analytical methods and release criteria is another key consideration. If the AOC platform has not yet established comprehensive release criteria—including the DAR distribution range, upper limit for free oligonucleotides, aggregate limits, and potency assay methods—then the path from clinical trials to commercialization remains long. A complete list of the CMC data package should be reviewed, rather than focusing solely on clinical data. When evaluating a platform transaction, the BD team should request to review
whether the platform can be repurposed across indications, whether there is clear evidence of tissue selectivity, whether the risks of long-term administration can be addressed, and whether analytical methods and release criteria suitable for regulatory communication have been established. These four questions constitute a comprehensive framework for assessing the maturity of AOC and small nucleic acid platforms. If the answers to all four questions are affirmative, the platform is ready to advance from early-stage clinical trials to late-stage development.If the answer to any of these questions is negative or uncertain, the platform remains in the proof-of-concept stage, and investment decisions must account for a corresponding risk premium.
Ask whether the platform can be repurposed across indications, whether there is clear evidence of tissue selectivity, whether the risks of long-term administration can be addressed, and whether analytical methods and release criteria suitable for regulatory communication have been established. The answers to these four questions collectively determine the distance between proof of concept and commercialization for AOC and small nucleic acid platforms. If the answer to any one of these questions is “no,” the platform remains in a high-risk early stage, and investment decisions must be adjusted accordingly.
5.3 Overview of New Cardiovascular Drugs: Which Projects Are Likely to Progress from Conference Buzz to Actual Products
Finally, by grouping GLP-1, AOC, small nucleic acids, and traditional cardiovascular drugs together and applying a five-dimensional framework, we can synthesize the assessments presented throughout this paper. These five dimensions are interrelated and must be evaluated holistically—if a project excels only in clinical evidence but has fundamental flaws in manufacturing feasibility or commercial reimbursement, its probability of progressing from conference buzz to an actual product will be significantly reduced.
| Evaluation Dimensions | Definition | GLP-1 (semaglutide/tirzopotide) | AOC/Small Nucleic Acids (Cardiovascular Indications) | Traditional Small-Molecule/Large-Molecule Cardiovascular Drugs |
| Clinical Evidence | Quantity and Quality of Positive Hard Endpoints | SELECT (MACE-20%) + FLOW (kidney) + STEP-HFpEF (heart failure) | DMD Phase 1/2 positive; cardiovascular indications still in the preclinical stage | Each has established standard-of-care status for specific indications, but incremental innovation is limited |
| Differentiation | Differences in efficacy, safety, and convenience compared to existing standard of care | Dosage frequency (weekly/monthly) + evidence for cardiovascular hard endpoints + multiple indications | Extrahepatic delivery (myocardium/CNS) represents a completely new delivery modality with no direct competitors | Requires combination with GLP-1 or siRNA, or identification of a differentiated patient subgroup |
| Manufacturing Feasibility | CMC maturity, production capacity, and supply chain stability | Peptide synthesis capacity is tight but scalable; the delivery device is the bottleneck | Coupling processes are complex, analytical methods are underdeveloped, and GMP-scale production is limited | Small-molecule and macromolecule manufacturing is mature, with low production capacity and supply chain risks |
| Regulatory Pathway | Progress in Communications with Regulatory Authorities and the Regulatory Environment | Cardiovascular indications have been approved, and the path for subsequent indications is clear | No dedicated AOC guidelines; case-by-case communication is required, resulting in high uncertainty | The regulatory pathway is well-established, but requirements for evidence of differentiation have increased |
| Commercial Reimbursement | Health insurance coverage, pricing levels, and patient copayment rates | Pricing is high, but expanded health insurance coverage for cardiovascular indications creates long-term reimbursement pressure | No commercial AOCs yet; pricing models have not been established | Amid generic drug price pressures, the innovation premium requires robust evidence to support it |
This five-dimensional evaluation table summarizes the findings of the full analysis.As shown in the table, GLP-1 is currently the only new class of cardiovascular drugs with relatively clear answers across all five dimensions—clinical evidence is supported by hard endpoints; differentiation is driven by advantages in dosing frequency and multiple indications; manufacturing feasibility, though challenging, is scalable; the regulatory pathway has been established; and commercial reimbursement is expanding from weight loss to cardiovascular prevention. Its primary uncertainties lie in long-term safety and rebound effects upon discontinuation.
This implies that AOC’s value in the cardiovascular field lies more in platform-level potential than in product-level certainty.To transition from conference buzz to actual products, AOC must overcome not only clinical validation but also the fact that, for AOC and small nucleic acids in cardiovascular indications, only the differentiation dimension currently offers clear advantages (with extrahepatic delivery being a new dimension), while the other four dimensions remain in early stages or are unclear. Establishing CMC maturity, regulatory pathways, and reimbursement models—each of these dimensions requires several years.
Traditional cardiovascular drugs face the most delicate situation: they are the most mature in terms of manufacturing feasibility and regulatory pathways, but face the greatest pressure regarding incremental innovation and differentiation in clinical evidence. Their path forward lies in finding opportunities in scenarios such as combination therapy, specific patient subgroups, and acute-phase treatment, rather than competing head-on with GLP-1 or nucleic acid drugs to secure an irreplaceable position.If developers of traditional drugs can identify complementary relationships between their pipelines and GLP-1/nucleic acid drugs at the ESC—rather than merely substitute relationships—they may be able to maintain their value within the new therapeutic landscape.
Action Recommendation: After the meeting, use the five-dimensional framework described above to score your own pipeline and the competitive assets you are monitoring (on a 1–5 point scale). The weighted average score across the five dimensions can serve as a reference for prioritizing projects. A low score in a particular dimension does not necessarily mean the project should be abandoned, but it does indicate that more resources need to be invested in that dimension for further validation.
When evaluating new cardiovascular drugs as a whole, participants should base their assessments on five key dimensions: clinical evidence, differentiation, manufacturability, regulatory pathways, and commercial reimbursement. These five dimensions form a comprehensive evaluation framework spanning the spectrum from science to commercialization, helping participants distinguish which projects will progress from conference buzz to actual products and which will remain merely at the conceptual stage.
| Evaluation Dimensions | Meets Standards | Signals of Failure | Post-Meeting Actions |
| Clinical Evidence | Phase III hard endpoint or strong signal from Phase II | Phase I or animal data only | Awaiting Phase III data |
| Differentiation | Unique mechanism/formulation/indication | No significant differences from existing drugs | Assessing the value of combination therapy |
| Manufacturing Feasibility | GMP-compliant manufacturing processes and release criteria in place | Process not finalized; analytical methods incomplete | CMC due diligence |
| Regulatory Pathway | Pre-IND/End-of-Phase 2 communications have taken place | No communication with regulatory authorities | Awaiting regulatory feedback |
| Commercial Payment | ICER within the reimbursement threshold | ICER exceeds the threshold; no health insurance coverage | Health Economics Assessment |
The clinical evidence dimension requires distinguishing between hard endpoints and surrogate endpoints.Hard endpoints for cardiovascular drugs include cardiovascular death, non-fatal myocardial infarction, non-fatal stroke, and hospitalization for heart failure. If the data presented for a project at the ESC conference only demonstrate surrogate endpoints—such as reductions in LDL-C or improvements in NT-proBNP—the assessment of its clinical value will require a longer validation period. The correlation between surrogate endpoints and hard endpoints must be established through specialized statistical validation and cannot be simply assumed.
Manufacturing feasibility is often overlooked, yet it determines whether a product can be delivered on time.A new cardiovascular drug that performs exceptionally well in clinical trials may face significant delays in its commercialization timeline if its formulation process cannot be scaled up, API production capacity is limited, or analytical methods are insufficient to support product release. When reviewing positive clinical data, attendees should simultaneously assess the project’s CMC maturity, which can typically be inferred from the formulation description in clinical trial registration information, the sponsor’s capacity announcements, and regulatory approval timelines.
Assessments of the commercial reimbursement dimension must take into account the healthcare policies of each country. For the same cardiovascular drug, coverage may be rapidly obtained through commercial insurance in the United States, while in Europe it must undergo an HTA (Health Technology Assessment) process, and in China it faces negotiations for inclusion in the National Medical Insurance Reimbursement List. Different reimbursement systems have varying levels of pricing tolerance for innovative drugs, which directly influences the design of a product’s global commercial strategy.
Summarizing the entire article using the five dimensions—clinical evidence, differentiation, manufacturing feasibility, regulatory pathway, and commercial payment—can make the piece more insightful than a typical pre-conference abstract. The assessment results for these five dimensions are also better suited for subsequent English SEO expansion—each dimension can be expanded into a separate in-depth analysis section, providing search engines with structured long-tail keyword coverage.
Authors should structure the entire article around these five dimensions: clinical evidence, differentiation, manufacturing feasibility, regulatory pathways, and commercial reimbursement. These dimensions are interrelated: the strength of clinical evidence influences the choice of regulatory pathway; the degree of differentiation affects willingness to pay; and manufacturing feasibility impacts product delivery capabilities and cost structure. When evaluating any project, attendees should consider all five dimensions simultaneously, rather than focusing on just one.

6. Pre-Conference Checklist: Turning the bio meeting 2026 into an Effective Information Screening Experience
With 33,000 attendees, four days, and hundreds of presentations, the ESC Congress 2026 can easily become a conference where you “listen to everything but remember nothing” without a clear information-filtering strategy. The efficiency of your attendance does not depend on how many presentations you attend, but rather on the questions you bring to them and how you process what you hear. The following three-stage checklist transforms your conference experience from “passively receiving information” to “actively filtering and evaluating evidence.”
Keep language concise, to the point, and actionable; avoid lengthy summaries. The following three sections provide specific guidelines for attending the conference, and each suggestion can be directly converted into an action list for attendees.
6.1 Pre-conference Screening: Focus on Three Categories—Hot Lines, GLP-1-Related Outcome Studies, and Signals Related to Cardiometabolic and Nucleic Acid Drugs
Scanning the entire ESC agenda from start to finish is the least efficient way to attend the conference. A more effective strategy is to first identify the three highest-priority content categories, mark them as “must-attend,” and then review the other sessions.
| Content Categories | Priority | Specific Screening Criteria | Location in the Agenda | Expected Outcomes |
| Hot Lines | Highest | All studies selected for Late-Breaking Science, with a special focus on cardiovascular outcomes, heart failure, and metabolic conditions | Hot Lines sessions on the first and second days of the conference | Primary endpoints, subgroup analyses, and safety updates for each study |
| Studies on GLP-1-related outcomes | High | SELECT extension analysis, SURPASS-CVOT update, STEP-HFpEF expanded data, FLOW subgroup | Metabolism and Heart Failure Sessions, Hot Lines | Latest Evidence on the Systemic Value of GLP-1 |
| Cardiovascular Metabolism + Nucleic Acid Therapeutics Signals | Medium to High | New Targets for Metabolic Syndrome, Heart Failure Drug Development, Early-Phase Clinical or Translational Research on Oligonucleotides/AOCs | Basic and Translational Medicine Session, Satellite Symposium | Early Signals and Technological Trends for Nucleic Acid Therapeutics in the Cardiovascular Field |
| Guideline Updates and Position Statements | Medium | Guideline updates and consensus document releases from various ESC working groups | Special Session Reports and Document Release Area | Potential Directions for Changes in Prescribing Practices |
| CMC and Regulatory Workshops | (CMC/BD Team) | CMC for Oligonucleotide Drugs, Regulatory Pathways for New Drugs, and Updates to Quality Standards | Satellite Sessions and Industry Exhibition Area | Updates on Manufacturing and Regulatory Aspects |
| Regular Keynote Presentations | Low (on-demand) | Select based on relevance to your pipeline | Each Session | Supplementary information; not considered core output |
The logic behind this table is: First, listen to content that most significantly impacts clinical practice, asset valuation, and R&D direction; then review the general topics. GLP-1-related research is the second priority, as it directly relates to data updates in the hottest current field. Signals on nucleic acid therapeutics are the third priority—although there may not be much data on cardiovascular-related nucleic acid therapeutics, any early signals are worth capturing because signals in this area are inherently scarce.“Hot Lines” are a must-listen and the top priority, as their release cycle is the fastest to impact the market.
Here’s a practical tip for screening: Go through the titles of all presentations on the agenda and categorize them using three labels—“Must-Attend,” “Optional,” and “Skip.”Limit “Must-Listen” sessions to no more than 15, “Optional” to no more than 20, and mark all others as “Skip.” This way, over the four-day conference, you’ll need to focus on no more than 35 presentations—each lasting about 20–30 minutes, for a total of approximately 15–20 hours—which is the maximum amount of information one person can effectively process during a four-day conference.
Attendees should focus on GLP-1-related outcome studies, as well as signals related to cardiovascular metabolism and nucleic acid therapeutics. The ESC Congress 2026 presents a vast agenda comprising hundreds of presentations, posters, and workshops. Attempting to cover everything is unrealistic. A more effective strategy is to first screen for the three categories of content most likely to impact clinical practice, asset valuation, and R&D direction: Hot Lines,
| Content Categories | Screening Criteria | Expected Outcomes | Recommended Time Allocation |
| Hot Lines | Cardiovascular drugs; Phase III data | Data that could influence guidelines | Prioritize attending all sessions |
| GLP-1 Outcome Studies | MACE subgroups; heart failure; CKD; safety | Assessment of the impact on prescribing behavior | Participate in at least 3–4 |
| Nucleic acid therapeutics/AOC signals | Extrahepatic delivery; new indications; CMC | Platform value assessment | 2–3 sessions + poster viewing |
| General topics | Related to own pipeline | Knowledge Updates | Flexible Scheduling |
Screening for Hot Lines is relatively straightforward—the ESC website announces the topics and study titles for Hot Lines in advance. Attendees should download the program before the conference, mark all Hot Lines sessions related to cardiovascular drugs, and review the published study protocols beforehand. This way, when the data is presented, they can quickly assess whether the results meet expectations, which data points exceed expectations, and which subgroup analyses warrant attention.
Screening for SELECT and GLP-1-related outcome studies requires greater attention to detail. In addition to the GLP-1 studies included in the Hot Lines, GLP-1-related data may also be presented in Late-Breaking Science, Clinical Science Updates, and special sessions.Attendees should search the agenda using keywords (semaglutide, tirzepatide, GLP-1, FLOW, STEP) to ensure they do not miss important sub-study data.
Screening for signals related to cardiovascular metabolism and nucleic acid therapeutics requires cross-agenda searches. Studies on nucleic acid therapeutics may be scattered across different sessions—the Basic Science session may feature reports on delivery technologies, the Translational Medicine session may include early clinical data on AOCs, and Late-Breaking Science may present major breakthroughs.Attendees should conduct a comprehensive search of the program using keywords such as “oligonucleotide,” “siRNA,” “AOC,” and “conjugate” to ensure they do not overlook nucleic acid drug-related content distributed across non-cardiovascular sessions.
Avoid the inefficient practice of reviewing the entire program. Prioritize content that most significantly impacts clinical practice, asset valuation, and R&D direction before moving on to general sessions. The ESC Congress program typically includes over 500 presentations and thousands of posters; attempting to cover everything is unrealistic. An effective strategy is to identify 3 to 5 content categories with the highest priority based on your role’s information needs and focus your efforts on gathering information from those categories.
6.2 During Q&A Sessions, Avoid Asking “What’s the Trend?”; Instead, Ask “Is the Data Sufficient to Change the Next Decision?”
At conferences like the ESC, the value of the Q&A session lies not in making your presence known, but in obtaining information not included in the presenter’s publicly shared data. Broad questions such as “What’s your take on the trends?” almost never yield useful answers—presenters will either offer a safe, generic response or simply repeat the conclusions from their presentation. A more effective approach is to focus your questions on specific decision-making dimensions.
| Dimensions for Asking Questions | Ineffective Questions (Avoid) | Effective Questions (Recommended) | Expected Information |
| Subgroup Data | “How effective is the treatment in different populations?” | “In the SELECT trial, was the reduction in MACE in the subgroup with BMI < 30 consistent with that of the overall population? Was there a statistically significant interaction?” | Quantitative Assessment of Subgroup Consistency |
| Long-term Safety | “What is the long-term safety profile?” | “In the extended follow-up of the SELECT trial, were there any changes in the incidence of pancreatitis and thyroid C-cell lesions? To what duration was the median follow-up extended?” | Specific data and timeline for the safety update |
| Mechanism and Clinical Benefits | “How does the drug work?” | “Do the cardiovascular benefits of GLP-1 stem primarily from weight loss or from direct cardiovascular protection? Are there any data from mediating analyses?” | Scientific Assessment of the Mechanism of Action |
| CMC and Supply Chain | “Is production capacity sufficient?” | “After the new indication is approved, will the planned expansion of dosing device production capacity be sufficient to meet the projected increase in patients? Are there alternative suppliers?” | Specific Plans for Manufacturing and Supply Chain |
| Regulatory Pathway | “When will approval be granted?” | “During pre-IND or end-of-Phase 2 meetings with the FDA/EMA, what evidence do the regulatory authorities require for the new indication?” | Specific content of regulatory communications |
| Differentiation Strategy | “How does it compare to competing products?” | “Are there plans for a head-to-head comparison with SGLT2 inhibitors in the HFpEF population? How do the differences in the two drugs’ targets translate into clinical differences?” | Scientific Basis for Competitor Comparisons |
The core principle of this table is: the more specific the question, the more meaningful the answer. Replacing “What do you make of the trend?” with “Is the data sufficient to change the next decision?” essentially transforms an open-ended inquiry about opinion into a factual confirmation with clear criteria for judgment. Presenters may not answer directly with “yes” or “no,” but their responses typically contain enough information for the questioner to make their own judgment.
Another questioning technique is to focus on data not presented in the report. If the presenter shows overall results but does not mention subgroup analyses, the questioner can ask for data on specific subgroups. If the report emphasizes efficacy but only briefly touches on safety, the questioner can ask about the incidence of specific adverse events. This “beyond-the-report” information often does not appear in conference abstracts or subsequent papers, yet it is the most meaningful for decision-making.
Asking questions on-site is a key channel for attendees to obtain non-public information. However, many attendees squander this opportunity by asking broad questions such as “What do you see as the trend?” or “What is the outlook?” A more valuable approach is to ask whether the data is sufficient to influence the next decision. Such questions force the presenter to provide specific assessments rather than make general statements.
| Types of Questions | Low-Value Questions | High-Value Questions | Expected Information Gained |
| Subgroup data | How effective is it in different populations? | Was the reduction in MACE statistically significant in the non-diabetic subgroup? | Determining the boundaries for indication expansion |
| Long-term safety | Is it safe in the long term? | What is the incidence of pancreatitis during follow-up of 3 years or more? How does it differ from the control group? | Assessing late-onset risks |
| Mechanism of Action | What is the mechanism? | How does clinical benefit correlate with the degree of weight loss? Is cardiovascular protection independent of weight loss? | Assessing added value |
| CMC Implications | Are there any manufacturing challenges? | If approved, what is the projected annual production capacity? Are there any supply bottlenecks? | Assessing the Commercialization Timeline |
Questions about subgroup data are the most practical. Research results presented at conferences typically show only the primary endpoints for the overall population, but subgroup analyses are often buried in posters or supplementary materials. By directly asking about data from key subgroups (non-diabetic patients, CKD, HFpEF, elderly) during the Q&A session, attendees can obtain the presenter’s own interpretation—which is far more informative than reading the paper afterward.
Questions regarding CMC and supply chain are often overlooked at clinical conferences but are crucial for pharmaceutical industry attendees. If a study presenter mentions that drug supply constraints have slowed enrollment, this suggests a production capacity bottleneck. Directly asking about projected annual production capacity and supply plans can help business development and supply chain teams evaluate collaboration or investment opportunities in advance.
Here is a set of reusable questions for readers: Are there subgroup data? Is there long-term safety data? Can the presenter explain the relationship between the mechanism of action and clinical benefit? Using these questions during the Q&A session allows you to obtain the presenter’s own assessment, which is more informative than reading the supplementary materials in the paper afterward. Will CMC or supply chain issues limit subsequent scale-up? These four questions apply to nearly all drug-related research presentations at the ESC. Attendees should
Provide readers with a set of reusable questions: Are there subgroup data? Is there long-term safety data? Can the relationship between the mechanism of action and clinical benefits be explained? Will CMC or supply chain issues limit subsequent scale-up? The rationale behind these four questions is as follows: subgroup data help determine the boundaries of indication expansion; long-term safety data help assess delayed risks; the relationship between the mechanism of action and clinical benefits helps determine differentiated value; and CMC and supply chain considerations help evaluate the commercialization timeline.
6.3 Post-Meeting Debriefing: Distinguish Between Three Key Areas—Clinical Validation, Platform Validation, and Commercial Scaling
After the meeting, the most common mistake attendees make is mixing all the information together, resulting in a vague “impression.” A more effective debriefing approach is to categorize the information into three groups, each corresponding to a different decision-making dimension.
| Dimensions of Debriefing | What to Look For | Evaluation Criteria | Post-Meeting Actions | Responsible Team |
| Clinical Validation | Is the endpoint hard? Is the patient population sufficiently broad? Is the safety profile acceptable? | Positive hard endpoint + consistency across populations + acceptable safety = Clinical Validation Passed | Update the internal medical evidence database and adjust the prescribing pathway assessment | Medical Affairs Team |
| Platform Validation | Is the delivery efficiency sufficient? Is the tissue selectivity clear? Are the CMC methods mature? | Quantifiable delivery to target tissues + Evidence of selectivity + Validated analytical methods = Preliminary platform validation passed | Update the platform technical assessment and adjust pipeline priorities | R&D + CMC Teams |
| Commercial Scale-Up | Can manufacturing keep up? Will payers accept it? How will the competitive landscape change? | Clear production capacity planning + Clear expectations for health insurance coverage + Differentiated competitive positioning = Feasible commercial scale-up pathway | Update the asset valuation model and adjust the BD strategy | BD + Commercial Team |
Clinical validation focuses on endpoints and patient populations; platform validation focuses on delivery and reusability; and commercial scaling focuses on manufacturing, reimbursement, and the competitive landscape. These three dimensions are not isolated but form a progressive sequence: clinical validation is a prerequisite for platform validation, and platform validation is the foundation for commercial scaling. If the clinical data is invalid, platform validation is meaningless; if platform validation fails, commercial scaling is out of the question.
A common misconception during post-mortems is equating positive clinical results directly with commercial success. A 20% reduction in MACE is clinically significant, but if production capacity cannot keep up, payers do not accept the pricing, or competitors launch a more differentiated solution within a year, this clinical advantage cannot be translated into commercial returns. The value of a three-dimensional post-mortem lies in preventing the misinterpretation of success in a single dimension as an overall victory.
The timing of the post-conference review is also crucial.It is recommended to complete the first round of rapid debriefing within 48 hours of the conference’s conclusion—at this point, memories are freshest, allowing for the rapid compilation of key data and preliminary assessments. Complete the second round of in-depth debriefing within one week, incorporating conference papers, peer reviews, and internal discussions to form a more systematic evaluation. Complete the third round of action implementation within two weeks—translate the debriefing conclusions into specific action items (such as pipeline adjustments, BD strategy updates, and revisions to medical materials), and assign responsible parties and deadlines.
Action Recommendation: Develop a three-dimensional post-meeting debriefing template after the meeting, using “Pass/Fail/To Be Verified” to rate each dimension. Assets that receive a “Pass” rating in all three dimensions can proceed to accelerated development or transaction evaluation; assets with a “To Be Verified” rating in any dimension require a tailored validation plan; and assets with a “Fail” rating in any dimension need to have their return on investment reassessed.
The post-meeting review should distinguish between three key areas: clinical validation, platform validation, and commercial scalability. These three terms can serve as the evaluation framework for the conclusion of this article. Clinical validation focuses on endpoints and patient populations—whether the study provides new hard endpoint data, which patient subgroups it covers, and whether the results are sufficient to alter guideline recommendations. Platform validation examines delivery and reusability—whether the AOC or small nucleic acid platform has demonstrated evidence of tissue selectivity and whether there is data supporting cross-indication transferability.Commercial scaling focuses on manufacturing, reimbursement, and the competitive landscape—whether the product can be delivered consistently, whether payers are willing to cover it, and how competitors’ pipelines are progressing.
| Dimensions for Review | Key Questions | Information Sources | Post-Meeting Actions |
| Clinical Validation | Hard Endpoints; Subgroups; Impact on Guidelines | Hot Lines Data; Abstract | Update on Medical Assessment |
| Platform Validation | Delivery efficiency; Tissue selectivity; Reusability | Poster; Investigator Communication | Technical Due Diligence Report |
| Commercial Scaling | Production capacity; payment; competition | Corporate Announcements; Analyst Reports | Update Business Plan |
The post-meeting review of clinical data should be completed within one week of the meeting. The team needs to compile all key data points from Hot Lines and Late-Breaking Science and evaluate them against the data interpretation framework established prior to the meeting. If data from a particular study significantly exceed or fall short of expectations, the team must analyze the reasons—whether due to differences in study design, patient selection, or endpoint definitions. This analysis helps the team establish more accurate expectations when evaluating similar studies in the future.
The post-meeting review for platform validation requires a longer timeframe. Data on tissue selectivity, safety signals, and CMC maturity assessments for the AOC and small nucleic acid platforms are typically obtained through post-meeting one-on-one discussions with investigators, literature searches, and corporate technical white papers. The BD team should complete a detailed due diligence report on the platform technology within one month after the meeting, including comparisons of delivery efficiency, assessments of analytical method coverage, and analyses of manufacturing feasibility.
The post-meeting review for commercial scaling should integrate conclusions from both the clinical and platform dimensions to reassess the strategic priorities of the product portfolio.If the data presented at the ESC confirms the long-term value of GLP-1 in the cardiovascular field, the company needs to reassess resource allocation for GLP-1-related projects in its pipeline. If the AOC platform demonstrates the feasibility of cross-tissue delivery, the company needs to decide whether to enter the AOC field through in-house development, collaboration, or acquisition. These strategic decisions should not be made at the conference itself but should be determined through collective discussion by management following a thorough review.
Clinical validation focuses on endpoints and patient populations; platform validation focuses on delivery and reusability; and commercial scaling focuses on manufacturing, reimbursement, and the competitive landscape. The debriefing for these three dimensions should be completed within different timeframes following the conference: clinical validation within one week, platform validation within one month, and commercial scaling within three months. This phased debriefing schedule ensures that both short-term signals and long-term assessments are thoroughly evaluated, avoiding hasty strategic decisions driven by the excitement of the conference.
These three points can serve as a framework for concluding the article. Clinical validation focuses on endpoints and patient populations—whether the study provided new hard endpoint data, covered new patient subgroups, and was sufficient to alter guideline recommendations. Platform validation focuses on delivery and reusability—whether the AOC or small nucleic acid platform demonstrated evidence of tissue selectivity and whether there is data supporting cross-indication transferability.For commercial scaling, examine manufacturing, reimbursement, and the competitive landscape—can the product be delivered consistently, are payers willing to cover it, and what is the progress of competing products in the pipeline?

7. bio meeting 2026 FAQ: Search-based Questions to Address When Writing About the ESC Congress
The following frequently asked questions address the information needs of search engine users. When answering, combine conference facts, industry insights, and the value of attending, rather than simply listing encyclopedic information.
7.1 When and where will the ESC Congress 2026 be held?
It will be held in Munich, Germany, from August 28 to 31, 2026. Official website: https://www.escardio.org/Congresses-Events/ESC-Congress. Based on 2025 data, the conference attracted over 33,300 attendees (including more than 29,300 healthcare professionals on-site and over 4,000 online participants) from 169 countries worldwide.The ESC Congress is one of the world’s largest and most academically influential cardiovascular medicine conferences. Each year, the ESC Congress 2026 (European Society of Cardiology Annual Congress 2026) features “Hot Lines” studies that directly influence the direction of updates to global cardiovascular diagnosis and treatment guidelines.
More than 33,300 attendees, including over 29,300 healthcare professionals on-site and more than 4,000 online participants.Attendance in 2026 is expected to remain on par with or slightly exceed that of 2025. Official information and registration details are available on the ESC website: https://www.escardio.org/Congresses-Events/ESC-CongressESC Congress 2026 will be held in Munich, Germany, from August 28 to 31, 2026.This is the European Society of Cardiology’s annual flagship academic conference and one of the most influential clinical medical conferences in the global cardiovascular field. The 2025 ESC Congress attracted participants from 169 countries
7.2 Why have GLP-1 drugs become a major highlight at cardiovascular conferences like the ESC?
The SELECT study demonstrated that GLP-1 receptor agonists can reduce the risk of MACE in overweight/obese individuals with cardiovascular disease. Breakthrough data announced during the “Hot Lines” session directly influence the guideline recommendations for GLP-1 drugs, payer reimbursement decisions, and the business development valuations of pharmaceutical companies.Additionally, while GLP-1 agents were initially developed as antidiabetic drugs for heart failure (STEP—semaglutide, tirzopotide), expanded data from recent cardiovascular outcomes trials—particularly in indications such as HFpEF and chronic kidney disease (FLOW)—have been continuously updated on the ESC platform, making it a core setting for assessing the systemic value of GLP-1. 20%.This data directly drove the FDA’s approval of Wegogy’s cardiovascular indication in March 2024, repositioning GLP-1 from a “weight-loss drug” to a “cardiovascular prevention drug.” As the premier platform for publishing cardiovascular outcome trials, the ESC annually presents
the SELECT study demonstrated a 20% reduction—the first hard-endpoint study to prove that GLP-1 receptor agonists have a cardiovascular protective effect independent of their glucose-lowering effects.Subsequently, the clinical positioning of GLP-1 drugs shifted from adjunctive antidiabetic agents to cardiovascular protective agents, and their prescribing pathways expanded from endocrinology to cardiology. The reduction in MACE risk is why GLP-1 drugs have become a major highlight at cardiovascular conferences such as the ESC—their cardiovascular outcome data have directly altered the treatment paradigm for cardiovascular and metabolic diseases.Semaglutide reduces the risk of major adverse cardiovascular events (MACE) in overweight or obese patients with a history of cardiovascular disease. As one of the leading global organizations for developing cardiovascular guidelines, the ESC’s presentation of GLP-1-related data at its annual congress directly influences adjustments to guideline recommendation levels, which in turn affect national health insurance reimbursement policies and prescribing practices.Furthermore, clinical research progress on GLP-1 in expanded indications—such as heart failure, chronic kidney disease, and non-alcoholic steatohepatitis (NASH)—has established it as a core drug class bridging the cardiovascular, metabolic, and renal disease fields.
7.3 Why Are AOCs and Small Nucleic Acid Drugs Worth Attention in the Cardiovascular Field?
AOCs (antibody-oligonucleotide conjugates) use antibodies to mediate the delivery of small nucleic acids (siRNA/PMO) to extrahepatic tissues (such as skeletal muscle, cardiac muscle, and the central nervous system), overcoming the limitation that traditional nucleic acid drugs can only target the liver.Novartis’s 2026 acquisition of Avidity Biosciences for approximately $12 billion reflects the industry’s high expectations for extrahepatic delivery platforms. For cardiovascular drug developers, the potential value of AOCs lies in:
① Pathogenic genes expressed in cardiac muscle tissue (such as MYBPC3 and MYH7) were previously inaccessible to siRNA; AOC may open up this therapeutic window,
② Local silencing of cardiovascular inflammatory factors may avoid the side effects of systemic immunosuppression
③ The ultra-long-acting nature of siRNA (administration once every six months to one year) is well-suited for the long-term management of chronic cardiovascular diseases
However, it is important to note that AOC is currently still in the preclinical stage for cardiovascular indications, and its myocardial delivery efficiency, tissue selectivity, long-term safety, and CMC maturity have yet to be validated. The platform’s value will ultimately need to be demonstrated through clinical data and product quality.
AOC and small nucleic acid drugs deserve attention in the cardiovascular field because breakthroughs in extrahepatic delivery technologies have opened up new therapeutic targets for cardiovascular diseases. The vast majority of currently approved small nucleic acid drugs utilize GalNAc-conjugation technology to achieve liver targeting, and are used to treat liver-related metabolic diseases such as hereditary transthyretin amyloidosis and hypercholesterolemia.However, many key pathological targets of cardiovascular disease—such as the TGF-beta pathway associated with myocardial fibrosis, ion channel subunits associated with arrhythmias, and signaling pathways associated with myocardial hypertrophy—are located in extrahepatic tissues, making selective intervention difficult with traditional small-molecule and large-molecule drugs.By combining the tissue-specific targeting of antibodies with the gene-silencing effects of oligonucleotides, AOC can theoretically achieve targeted delivery to extrahepatic tissues such as cardiac and skeletal muscle. Early clinical data from AOC 1001 in DMD patients have preliminarily validated the feasibility of the AOC platform in skeletal muscle.If this platform can be translated to cardiac muscle tissue, it will provide entirely new therapeutic strategies for cardiovascular diseases—such as cardiac fibrosis, hereditary cardiomyopathies, and arrhythmias—for which effective targeted treatments are currently lacking. However, attendees should remain cautious: the AOC platform’s tissue selectivity, long-term safety, and CMC maturity still require further validation through clinical and process data, and the translational pathway from DMD to cardiovascular applications has not yet been clinically confirmed. Avidity Biosciences’
7.4 What should pharmaceutical companies or CDMOs focus on at this conference?
Pharmaceutical companies and CDMOs attending the ESC Congress 2026 should shift their focus from “who presented impressive results” to “what development needs these results will generate.” Specifically:
① Positive results for new GLP-1 indications will drive manufacturing needs such as formulation specification expansion, production capacity expansion, and delivery device upgrades
② Early signals for AOCs and small nucleic acids, if they emerge, will drive CDMO demand for conjugation process development, analytical characterization methods, and GMP production capacity
③ Traditional cardiovascular drugs, under pressure from GLP-1 agonists, will require evidence of differentiation, which may spur clinical development of combination therapies and demand for compound formulations
④ Updates to guidelines for oligonucleotide drugs and review criteria for cardiovascular drugs discussed at regulatory workshops will directly impact development strategies and submission pathways
Make investment decisions based on the peak sentiment following the release of Hot Lines. CMC teams should pay particular attention to CMC workshops and supply chain company developments surrounding the conference, translating clinical signals into development challenges for manufacturing and quality systems. BD teams should distinguish between “validated assets” and “platforms driven by sentiment” to avoid
pharmaceutical companies or CDMOs participating in GLP-1-related clinical trials may generate new formulation development needs—for example, data from CKD subgroups may spur the development of lower-dose formulations, while clinical progress in AOCs and small nucleic acids will impact demand for CDMO services. If an AOC platform demonstrates preliminary evidence of cross-tissue delivery, CDMOs need to assess whether to invest in conjugation process capabilities and oligonucleotide synthesis capacity.Third, which regulatory trends will alter requirements for analytical method development—statements by regulatory representatives and content from satellite sessions at the ESC often reveal the direction of future guidance updates. The key focus for the 2026 ESC Congress should shift from clinical data to operational-level assessments. Specifically, attention should be paid to the following areas: First, which CMC teams need to plan ahead.Fourth, which competitor data will influence the prioritization of one’s own pipeline? If competitors release better-than-expected data at the ESC, companies will need to reassess the differentiation potential and time window for their own projects. CDMO companies, meanwhile, should attend the conference from the perspective of client needs: paying attention to which pharmaceutical companies’ clinical projects have made progress at the ESC, and what process development, analytical methods, and GMP manufacturing services these projects will require as they advance to the next phase of development.HFpEF data may drive the evaluation of combination therapies involving GLP-1 and standard heart failure treatments. Second, which





